

A well-developed knowledge of clinical microbiology is
critical for the practicing physician in any medical field.
Bacteria, viruses, and protozoans have no respect for
the distinction between ophthalmology, pediatrics,
trauma surgery, or geriatric medicine. As a physician
you
will be faced daily with the concepts of microbial
disease and antimicrobial therapy. Microbiology is one
of the few courses where much of the "minutia" is regu-
larly used by the practicing physician.
This book attempts to facilitate the learning of mi-
crobiology by presenting the information in a clear and
entertaining manner brimming with memory aids.
Our approach has been to:
1) Write in a conversational style for rapid assimi-
lation.
2) Include numerous figures serving as "visual mem-
ory
tools" and summary charts at the end of each chap-
ter. These can be used for "cram sessions" after the
concepts have been studied in the text.
3) Concentrate more on clinical and infectious dis-
ease issues that are both interesting and vital to the ac-
tual practice of medicine.
Preface
D
4) Create a conceptual, organized approach to the or-
ganisms studied so the student relies less on memory
and more on logical pathophysiology.
The text has been updated to include current infor-
mation on rapidly developing topics, such as HIV and
AIDS (vaccine efforts and all the new anti-HIV medica-
tions), Ebola virus, Hantavirus, E. coli outbreaks, Mad
Cow Disease, and brand-new antimicrobial antibiotics.
The mnemonics and cartoons in this book do not in-
tend disrespect for any particular patient population or
racial or ethnic group but are solely presented as mem-
ory devices to assist in the learning of a complex and im-
portant medical subject.
We welcome suggestions for future editions.
MARK GLADWIN, MD
BILL TRATTLER, MD

CONTENTS
Preface
v
PART 1
1
1
BACTERIAL TAXONOMY
1
2
CELL STRUCTURES, VIRULENCE FACTORS, and TOXINS
8
3
BACTERIAL SE( GENETICS
16
GRAM-POSITIVE BACTERIA
22
4
STREPTOCOCCUS
22
5
STAPHYLOCOCCUS
31
6
BACILLUS and CLOSTRIDIUM (SPORE-FORMING RODS)
38
7
CORYNEBACTERIUM and LISTERIA (NON-SPORE-FORMING RODS)
45
GRAM-NEGATNE BACTERIA
49
8
NEISSERIA
49
9
THE ENTERICS
54
10
HAEMOPHILUS, BORDETELLA, and LEGIONELLA
68
11
YERSINIA, FRANCISELLA, BRUCELLA, and PASTEURELLA
73
12
CHLAMYDIA, RICKETTSIA, and FRIENDS
78
13
SPIROCHETES
91
ACID-FAST BACTERIA
102
14
MYCOBACTERIUM
102
BACTERIA WITHOUT CELL WALLS
111
15
MYCOPLASMA
111
ANTI-BACTERIAL MEDICATIONS
114
16
PENICILLIN FAMILY ANTIBIOTICS
114
17
ANTI-RIBOSOMAL ANTIBIOTICS
125
18
ANTI-TB and ANTI-LEPROSY ANTIBIOTICS
133
19
MISCELLANEOUS ANTIBIOTICS
139
PART 2. FUNGI
144
20
THE FUNGI
144
21
ANTI-FUNGAL MEDICATIONS
155
PART 3
161
22
VIRAL REPLICATION and TAXONOMY
161
23
ORTHOMYXO and PARAMYXOVIRIDAE
172
24
HEPATITIS VIRIDAE
180
25
RETROVIRIDAE, HIV, and AIDS
190
26
HERPESVIRIDAE
204
27
REST OF THE DNA VIRUSES
209
28
REST OF THE RNA VIRUSES
214
29
ANTI-VIRAL MEDICATIONS
224
PART 4. PARASITES
231
30
PROTOZOANS
231
31
HELMINTHS
248
vi

PART 5.
32
PRIONS (contributing author: Hans Henrik Larsen, M.D.)
265
PART 6.
33
ANTIMICROBIAL RESISTANCE: ONE STEP TOWARD THE POST-ANTIBIOTIC ERA?
(contributing author: Earnest Alexander, Pharm.D.)
269
BIOTERRORISM DEFENSE UPDATES:
http://www.medmaster.netBioterrorismDefense.html
VERY STRANGE CRITTERS
265


A well-developed knowledge of clinical microbiology is
critical for the practicing physician in any medical field.
Bacteria, viruses, and protozoans have no respect for
the distinction between ophthalmology, pediatrics,
trauma surgery, or geriatric medicine. As a physician
you
will be faced daily with the concepts of microbial
disease and antimicrobial therapy. Microbiology is one
of the few courses where much of the "minutia" is regu-
larly used by the practicing physician.
This book attempts to facilitate the learning of mi-
crobiology by presenting the information in a clear and
entertaining manner brimming with memory aids.
Our approach has been to:
1) Write in a conversational style for rapid assimi-
lation.
2) Include numerous figures serving as "visual mem-
ory
tools" and summary charts at the end of each chap-
ter. These can be used for "cram sessions" after the
concepts have been studied in the text.
3) Concentrate more on clinical and infectious dis-
ease issues that are both interesting and vital to the ac-
tual practice of medicine.
Preface
D
4) Create a conceptual, organized approach to the or-
ganisms studied so the student relies less on memory
and more on logical pathophysiology.
The text has been updated to include current infor-
mation on rapidly developing topics, such as HIV and
AIDS (vaccine efforts and all the new anti-HIV medica-
tions), Ebola virus, Hantavirus, E. coli outbreaks, Mad
Cow Disease, and brand-new antimicrobial antibiotics.
The mnemonics and cartoons in this book do not in-
tend disrespect for any particular patient population or
racial or ethnic group but are solely presented as mem-
ory devices to assist in the learning of a complex and im-
portant medical subject.
We welcome suggestions for future editions.
MARK GLADWIN, MD
BILL TRATTLER, MD

CONTENTS
Preface
v
PART 1
1
1
BACTERIAL TAXONOMY
1
2
CELL STRUCTURES, VIRULENCE FACTORS, and TOXINS
8
3
BACTERIAL SE( GENETICS
16
GRAM-POSITIVE BACTERIA
22
4
STREPTOCOCCUS
22
5
STAPHYLOCOCCUS
31
6
BACILLUS and CLOSTRIDIUM (SPORE-FORMING RODS)
38
7
CORYNEBACTERIUM and LISTERIA (NON-SPORE-FORMING RODS)
45
GRAM-NEGATNE BACTERIA
49
8
NEISSERIA
49
9
THE ENTERICS
54
10
HAEMOPHILUS, BORDETELLA, and LEGIONELLA
68
11
YERSINIA, FRANCISELLA, BRUCELLA, and PASTEURELLA
73
12
CHLAMYDIA, RICKETTSIA, and FRIENDS
78
13
SPIROCHETES
91
ACID-FAST BACTERIA
102
14
MYCOBACTERIUM
102
BACTERIA WITHOUT CELL WALLS
111
15
MYCOPLASMA
111
ANTI-BACTERIAL MEDICATIONS
114
16
PENICILLIN FAMILY ANTIBIOTICS
114
17
ANTI-RIBOSOMAL ANTIBIOTICS
125
18
ANTI-TB and ANTI-LEPROSY ANTIBIOTICS
133
19
MISCELLANEOUS ANTIBIOTICS
139
PART 2. FUNGI
144
20
THE FUNGI
144
21
ANTI-FUNGAL MEDICATIONS
155
PART 3
161
22
VIRAL REPLICATION and TAXONOMY
161
23
ORTHOMYXO and PARAMYXOVIRIDAE
172
24
HEPATITIS VIRIDAE
180
25
RETROVIRIDAE, HIV, and AIDS
190
26
HERPESVIRIDAE
204
27
REST OF THE DNA VIRUSES
209
28
REST OF THE RNA VIRUSES
214
29
ANTI-VIRAL MEDICATIONS
224
PART 4. PARASITES
231
30
PROTOZOANS
231
31
HELMINTHS
248
vi

PART 5.
32
PRIONS (contributing author: Hans Henrik Larsen, M.D.)
265
PART 6.
33
ANTIMICROBIAL RESISTANCE: ONE STEP TOWARD THE POST-ANTIBIOTIC ERA?
(contributing author: Earnest Alexander, Pharm.D.)
269
BIOTERRORISM DEFENSE UPDATES:
http://www.medmaster.netBioterrorismDefense.html
VERY STRANGE CRITTERS
265
CHAPTER 1. BACTERIAL TAXONOMY
All organisms have a name consisting of two parts: the
genus followed by the species (i.e., Homo sapiens). Bac-
teria have been grouped and named primarily on their
morphological and biochemical/metabolic differences.
However, bacteria are now also being classified accord-
ing to their immunologic and genetic characteristics.
This chapter focuses on the Gram stain, bacterial mor-
phology, and metabolic characteristics, all of which en-
able the clinician to rapidly determine the organism
causing a patient's infection.
GRAM STAIN
Because bacteria are colorless and usually invisible to
light microscopy, colorful stains have been developed to
visualize them. The most useful is the Gram stain,
which separates organisms into 2 groups: gram-positive
bugs and gram-negative bugs. This stain also allows the
clinician to determine whether the organism is round or
rod-shaped.
For any stain you must first smear the substance to
be stained (sputum, pus, etc.) onto a slide and then heat
it to fix the bacteria on the slide.
There are 4 steps to the Gram stain:
1) Pour on crystal violet stain (a blue dye) and wait
60 seconds.
2) Wash off with water and flood with iodine solu-
tion. Wait 60 seconds.
3) Wash off with water and then "decolorize" with
95% alcohol.
4) Finally, counter-stain with safranin (a red dye).
Wait 30 seconds and wash off with water.
When the slide is studied microscopically, cells that
absorb the crystal violet and hold onto it will appear
blue. These are called gram-positive organisms. How-
ever, if the crystal violet is washed off by the alcohol,
these cells will absorb the safranin and appear red.
These are called gram-negative organisms.
Gram-positive = BLUE
I' m positively BLUE over you!!
Gram-negative = RED
No (negative) RED commies!!
The different stains are the result of differences in the
cell walls of gram-positive and gram-negative bacteria.
PART 1. BACTERIA
1
DISACCHARIDE
AMINO ACIDS
Figure 1-1
Both gram-positive and gram-negative organisms
have more than 1 layer protecting their cytoplasm and
nucleus from the extracellular environment, unlike an-
i mal cells, which have only a single cytoplasmic mem-
brane composed of a phospholipid bilayer. The layer just
outside the bacterial cytoplasmic membrane is the pep-
tidoglycan layer or cell wall. It is present in both
gram-positive and gram-negative organisms.
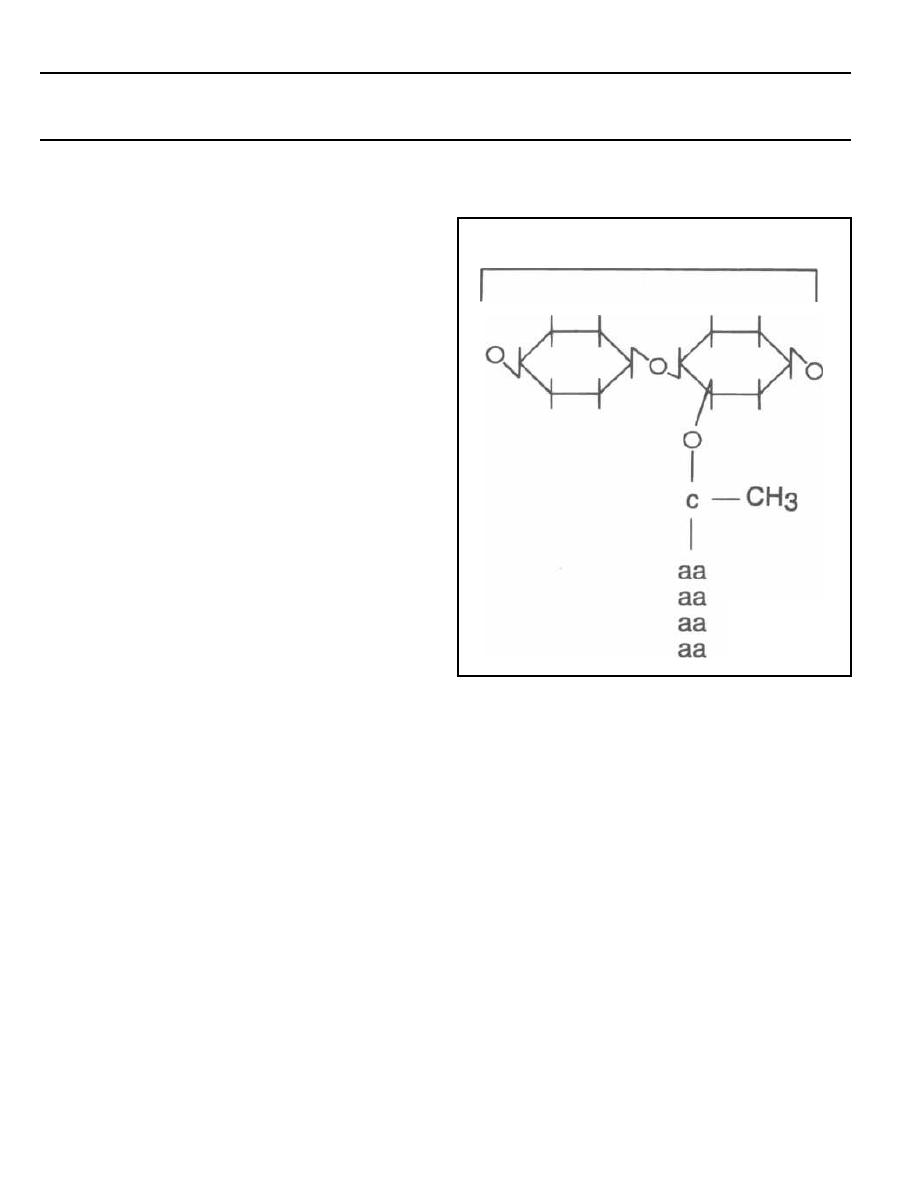
Fig. 1-1.
The peptidoglycan layer or cell wall is
composed of repeating disaccharides with 4 amino acids
in a side chain extending from each disaccharide.
Fig. 1-2.
The amino-acid chains of the peptidoglycan
covalently bind to other amino acids from neighboring
chains. This results in a stable cross-linked structure.
The enzyme that catalyzes the formation of this linkage
is called transpeptidase and is located in the inner cy-
toplasmic membrane. The antibiotic penicillin binds to
and inhibits this enzyme. For this reason the enzyme is
also called penicillin binding protein (see page 114).
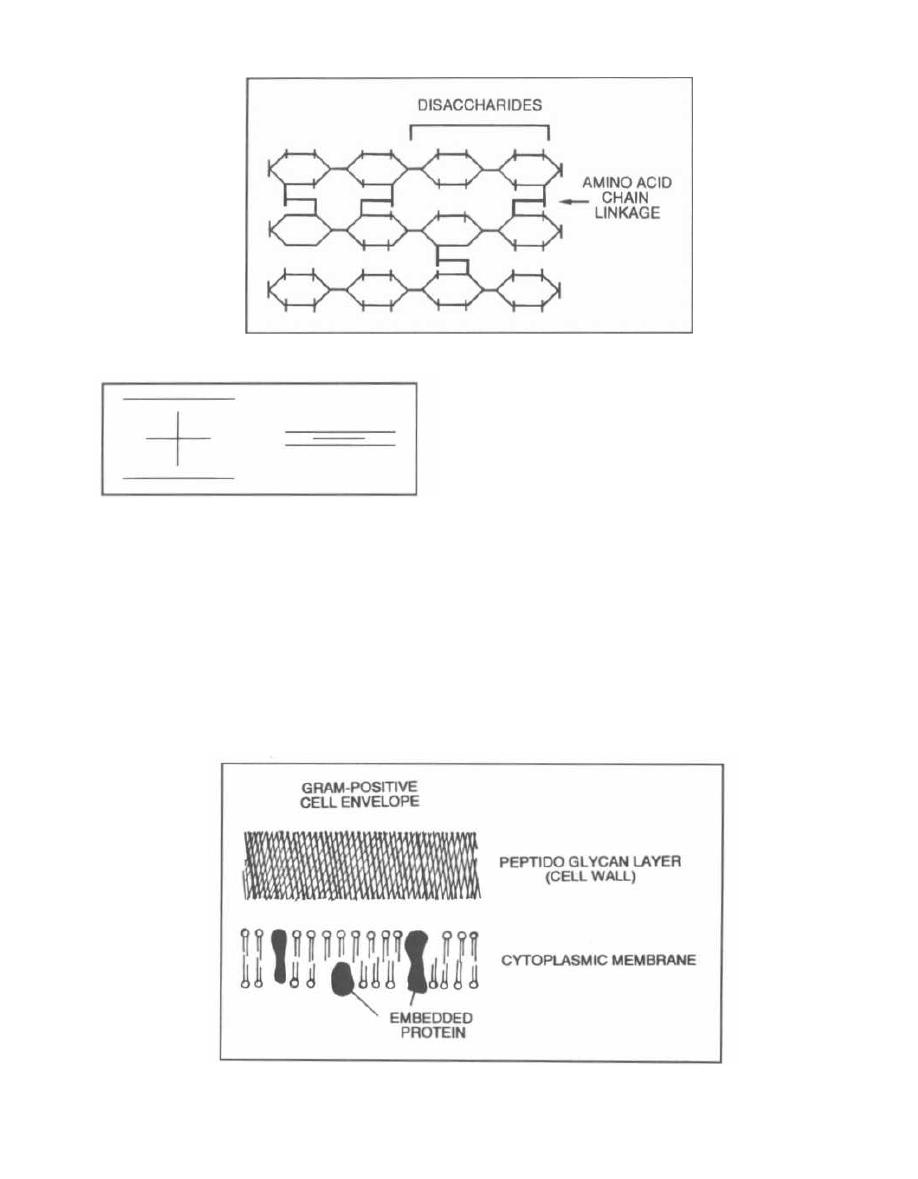
Figure 1-2
Figure 1-3
Fig. 1-3. The gram-positive cell wall is very thick and
has extensive cross-linking of the amino-acid side
chains. In contrast, the gram-negative cell wall is very
thin with a fairly simple cross-linking pattern.
Fig. 1-4. The gram-positive cell envelope has an outer
cell wall composed of complex cross-linked peptidogly-
can, teichoic acid, polysaccharides, and other proteins.
The inner surface of the cell wall touches the cytoplas-
mic membrane. The cytoplasmic membrane contains
proteins that span the lipid bilayer. The bacterial cyto-
Figure 1-4
CHAPTER 1. BACTERIAL TAXONOMY
plasmic membrane (unlike that of animals) has no cho-
lesterol or other sterols.
An important polysaccharide present in the gram-
positive cell wall is teichoic acid. It acts as an antigenic
determinant, so it is important for serologic identifica-
tion of many gram-positive species.
Fig. 1-5. The gram-negative cell envelope has S layers,
not including the periplasmic space. Like gram-positive
bacteria, it has 1) a cytoplasmic membrane surrounded
by 2) a peptidoglycan layer. 3) In addition, a gram-
negative cell has a unique outer cell membrane.
The inner cytoplasmic membrane (as in gram-positive
bacteria) contains a phospholipid bilayer with embed-
ded proteins. Gram-negative bacteria have a periplas-
mic space between the cytoplasmic membrane and an
extremely thin peptidoglycan layer. This periplasmic
space is filled with a gel that contains proteins and en-
zymes. The thin peptidoglycan layer does not contain
teichoic acid, although it does have a small helical
2
CHAPTER 1. BACTERIAL TAXONOMY
Figure 1-5
Figure 1-6
3
lipoprotein called murein lipoprotein. This lipopro-
tein is important because it originates from the peptido-
glycan layer and extends outward to bind the unique
third outer membrane. This last membrane is similar to
other cell membranes in that it is composed of two lay-
ers of phospholipid (bilayer) with hydrophobic tails in
the center. What makes it unique is that the outermost
portion of the bilayer contains lipopolysaccharide (LPS).
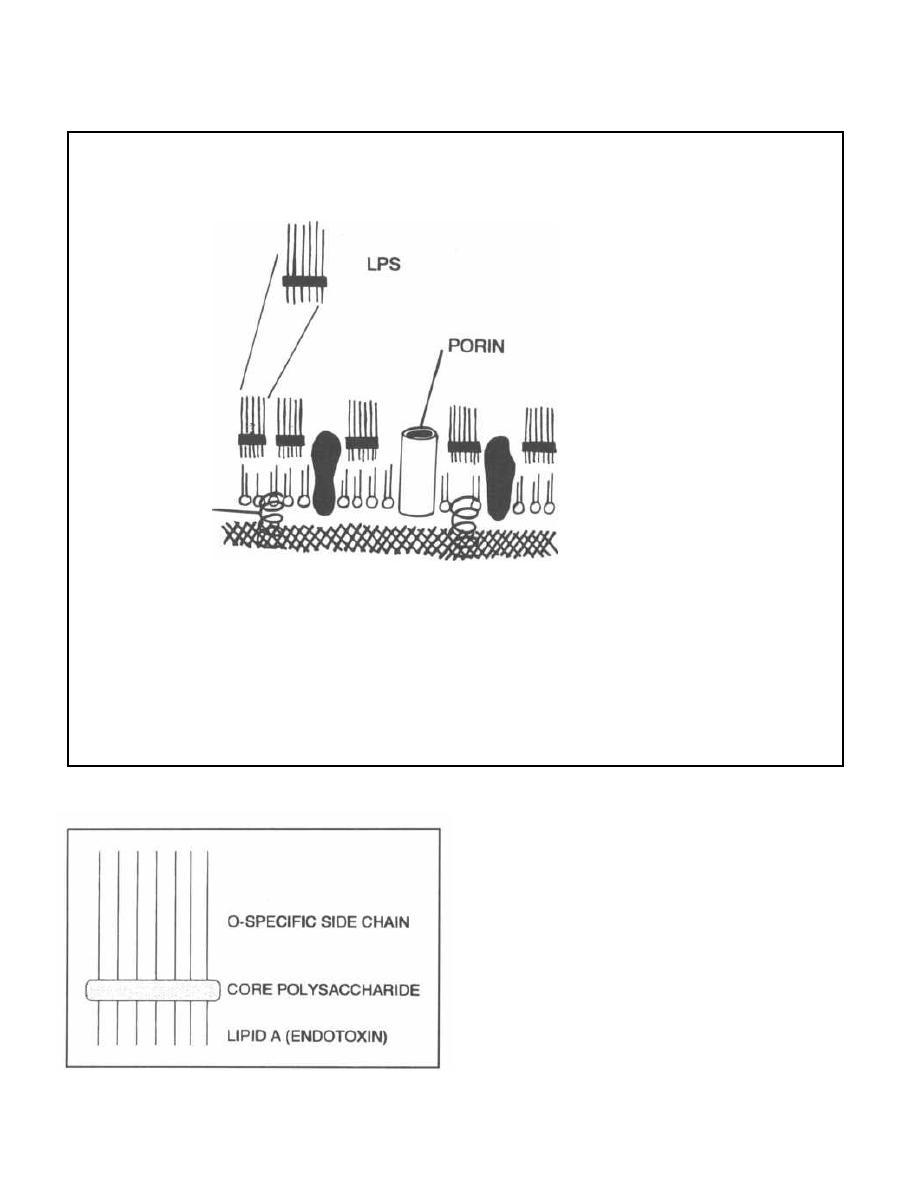
Fig. 1-6.
Lipopolysaccharide (LPS) is composed of 3
covalently linked components:
1) Outer carbohydrate chains of 1-50 oligosaccha-
ride units that extend into the surrounding media.
These differ from one organism to another and are anti-
genic determinants. This part is called the 0-specific
MUREIN
LIPOPROTEIN
GRAM-NEGATIVE
CELL ENVELOPE
PERIPLASMIC SPACE
EMBEDDED
PROTEINS
OUTER MEMBRANE
PEPTIDOGLYCAN LAYER
(CELL WALL)
CYTOPLASMIC MEMBRANE

side chain or the 0-antigen. Think of O for Outer to
help remember this.
2) The center part is a water soluble core polysac-
charide.
3) Interior to the core polysaccharide is the third
component, lipid A, which is a disaccharide with mul-
tiple fatty acid tails reaching into the membrane. Lipid
A is toxic to humans and is known as the gram-negative
endotoxin. When bacterial cells are lysed by our effi-
ciently working immune system, fragments of mem-
brane containing lipid A are released into the
circulation, causing fever, diarrhea, and possibly fatal
endotoxic shock (also called septic shock).
Embedded in the gram-negative outer membrane are
porin proteins, which allow passage of nutrients. These
are also unique to gram-negative organisms.
What does this mean clinically?
The differences between gram-positive and gram-
negative organisms result in varied interactions with
the environment. The gram-positive thickly meshed
peptidoglycan layer does not block diffusion of low mol-
ecular weight compounds, so substances that damage
the cytoplasmic membrane (such as antibiotics, dyes,
and detergents) can pass through. However, the gram-
negative outer lipopolysaccharide-containing cell mem-
brane blocks the passage of these substances to the
peptidoglycan layer and sensitive inner cytoplasmic
membrane. Therefore, antibiotics and chemicals that
attempt to attack the peptidoglycan cell wall (such as
penicillins and lysozyme) are unable to pass through.
Interestingly, the crystal violet stain used for Gram
staining is a large dye complex that is trapped in the
thick, cross-linked gram-positive cell wall, resulting in
the gram-positive blue stain. The outer lipid-containing
cell membrane of the gram-negative organisms is par-
CHAPTER 1. BACTERIAL TAXONOMY
Figure 1-7 DIFFERENCES BETWEEN GRAM-
-POSITIVE AND GRAM-NEGATIVE ORGANISMS
4
dally dissolved by alcohol, thus washing out the crystal
violet and allowing the safranin counterstain to take.
Fig. 1-7.
Summary of differences between gram-
positive and gram-negative bacteria.
BACTERIAL MORPHOLOGY
Bacteria have 4 major shapes:
1) Cocci: spherical.
2) Bacilli:
rods.
Short bacilli are called
coc-
cobacilli.
3) Spiral forms: comma-shaped, S-shaped, or spi-
ral-shaped.
4) Pleomorphic: lacking a distinct shape (like jello).
The different shaped creatures organize together into
more complex patterns, such as pairs (diplococci), clus-
ters, strips, and single bacteria with flagella.
Fig. 1-8.
Bacterial morphology.
SO, WHAT ARE THE NAMES?!!!!
Gram-Positive
Start by remembering that there are 6 classic gram-
positive bugs that cause disease in humans, and basi-
cally every other organism is gram-negative.
Of the gram-positives, 2 are cocci, and the other 4 are
rod-shaped (bacilli).
The 2 gram-positive cocci both have the word coccus
in their names:
1) Streptococcus forms strips of cocci.
2) Staphylococcus forms clusters of cocci.
Two of the 4 gram-positive rods produce spores
(spheres that protect a dormant bacterium from the
harsh environment). They are:
Gram-Positive Cells
Gram-Negative Cells
2 Layers:
1. Inner cytoplasmic membrane
2. Outer thick peptidoglycan layer
(60-100% peptidoglycan)
3 Layers:
1. Inner cytoplasmic membrane
2. Thin peptidoglycan layer
(5- 10% peptidoglycan)
3. Outer membrane with
lipopolysaccharide (LPS)
Low lipid content
High lipid content
NO endotoxin (except
Listeria
monocytogenes)
Endotoxin (LPS) - lipid A
NO periplasmic space
Periplasmic space
NO porin channel
Porin channel
Vulnerable to lysozyme and
penicillin attack
Resistant to lysozyme and
penicillin attack

COCCI
BACILLI or RODS
SPIRAL
COMMA
S-SHAPED
PLEOMORPHIC
CLUSTERS
STRIPS
DIPLOCOCCI
WITH FLAGELLA
Figure 1-8
3) Bacillus
4) Clostridium
The last 2 gram-positive rods do not form spores:
5) Corynebacterium
6) Listeria,
which surprisingly has endotoxin-sur-
prising because ALL other organisms with endotoxin
are gram-negative.
CHAPTER 1. BACTERIAL TAXONOMY
5
Gram-Negative
Of the gram-negative organisms, there is only one
group of gram-negative cocci. It is actually a diplococcus
(looks like 2 coffee beans kissing):
Neisseria.
There is also just 1 group of spiral-shaped organisms:
the Spirochetes. This group includes the bacterium
Tre-
ponema pallidum,
which causes syphilis.
The rest are gram-negative rods or pleomorphic.
Exceptions:
1) Mycobacteria are weakly gram-positive but
stain better with a special stain called the acid-fast
stain (See Chapter 14). This special group includes or-
ganisms that cause tuberculosis and leprosy.
2) Spirochetes have a gram-negative cell wall
but are too small to be seen with the light microscope
and so must be visualized with a special darkfield
microscope.
3) Mycoplasma do not have a cell wall. They only
have a simple cell membrane, so they are neither gram-
positive nor gram-negative.
Fig.
1-9.
Summary of morphological differences
among the bacteria.
CYTOPLASMIC STRUCTURES
Bacterial DNA usually consists of a single circle of
double-stranded DNA. Smaller adjacent circles of
double-stranded DNA are called plasmids; they often
contain antibiotic resistance genes. Ribosomes are
composed of protein and RNA and are involved in the
translation process, during the synthesis of proteins.
Bacteria, which are procaryotes, have smaller ribo-
somes (70S) than animals (80S), which are eucaryotes.
Bacterial ribosomes consist of 2 subunits, a large sub-
unit
( 50S)
and a small subunit (30S). These numbers re-
late to the rate of sedimentation. Antibiotics, such as
erythromycin and tetracycline, have been developed
that attack like magic bullets. They inhibit protein syn-
thesis preferentially at the bacterial ribosomal subunits
while leaving the animal ribosomes alone. Erythromy-
cin works at the 50S subunit, while tetracycline blocks
protein synthesis at the 30S subunit.
METABOLIC CHARACTERISTICS
Bacteria can be divided into groups based on their
metabolic properties. Two important properties include:
1) how the organism deals with oxygen, and 2) what the
organism uses as a carbon and energy source. Other
properties include the different metabolic end-products
that bacteria produce such as acid and gas.

CHAPTER 1. BACTERIAL TAXONOMY
Figure 1-9 MORPHOLOGICAL DIFFERENCES AMONG THE BACTERIA
Oxygen
How bacteria deal with oxygen is a major factor in
their classification. Molecular oxygen is very reactive,
and when it snatches up electrons, it can form hydrogen
peroxide (H2O2), superoxide radicals (02i, and a hy-
droxyl radical (OH
-
). All of these are toxic unless bro-
ken down. In fact, our very own macrophages produce
these oxygen radicals to pour over bacteria. There are 3
enzymes that some bacteria possess to break down
these oxygen products:
1) Catalase breaks down hydrogen peroxide in the
following reaction:
6
3) Superoxide dismutase breaks down the super-
oxide radical in the following reaction:
2) Peroxidase also breaks down hydrogen peroxide.
Bacteria are classified on a continuum. At one end are
those that love oxygen, have all the preceding protective
enzymes, and cannot live without oxygen. On the oppo-
site end are bacteria which have no enzymes and pretty
much kick the bucket in the presence of oxygen:
1) Obligate aerobes: These critters are just like us
in that they use glycolysis, the Krebs TCA cycle, and the
electron transport chain with oxygen as the final elec-
tron acceptor. These guys have all the above enzymes.
2) Facultative anaerobes: Don't let this name fool
you! These bacteria are aerobic. They use oxygen as an
electron acceptor in their electron transfer chain and
MORPHOLOGY
GRAM-POSITIVE
GRAM-NEGATIVE
Circular (Coccus)
Streptococcus
Staphylococcus
Neisseria
Rod (Bacillus)
Corynebacterium
Listeria
Bacillus
Clostridium
Mycobacterium (acid-fast)
ENTERICS (live in the GI tract):
E
scherichia coli
S
higella
S
almonella
• Y
ersinia
K
lebsiella
P
roteus
E
nterobacter
Serratia
V
ibrio
C
ampylobacter
H
elicobacter
P
seudomonas
'BacteroIdes (anaerobic)
Haemophilus
Bordetella
Legionella
Yersinia
Francisella
Brucella
Pasteurella
Gardnerella
Spiral
Spirochetes:
T
reponema
B
orrelia
L
eptospira
Branching filamentous growth
(like fungi)
Actinomyces (anaerobic)
Nocardia (partially acid-fast)
Pleomorphic
Chlamydia
Rickettsiae
No cell wall
Mycoplasma
CHAPTER 1. BACTERIAL TAXONOMY
*Chlamydia
and Rickettsia do not have the metabolic machinery to utilize oxygen. They are energy parasites,
and must steal their host's ATP.
Figure 1-10 OXYGEN SPECTRUM
have catalase and superoxide dismutase. The only dif-
ference is that they can grow in the absence of oxygen
by using fermentation for energy. Thus they have the
faculty to be anaerobic but prefer aerobic conditions.
Phis is similar to the switch to anaerobic glycolysis that
human muscle cells undergo during sprinting.
3) Microaerophilic bacteria (also called aero-
tolerant anaerobes): These bacteria use fermentation
and have no electron transport system. They can toler-
ate low amounts of oxygen because they have superox-
ide dismutase (but they have no catalase).
4) Obligate anaerobes: These guys hate oxygen
and have no enzymes to defend against it. When you are
working on the hospital ward, you will often draw blood
for culture. You will put the blood into 2 bottles for
growth. One of these is an anaerobic growth media with
no oxygen in it!
Fig. 1-10.
The oxygen spectrum of the major bacterial
groups.
Carbon and Energy Source
Some organisms use light as an energy source (pho-
totrophs), and some use chemical compounds as an en-
ergy source (chemotrophs). Of the organisms that use
chemical sources, those that use inorganic sources, such
as ammonium and sulfide, are called autotrophs. Oth-
ers use organic carbon sources and are called het-
erotrophs. All the medically important bacteria are
chemoheterotrophs because they use chemical and
organic compounds, such as glucose, for energy.
Fermentation (glycolysis) is used by many bacteria
for oxygen metabolism. In fermentation, glucose is bro-
ken down to pyruvic acid, yielding ATP directly. There
are different pathways for the breakdown of glucose to
pyruvate, but the most common is the Embden-
Meyerhof pathway. This is the pathway of glycolysis
that we have all studied in biochemistry. Following fer-
mentation the pyruvate must be broken down, and the
different end products formed in this process can be
used to classify bacteria. Lactic acid, ethanol, propionic
acid, butyric acid, acetone, and other mixed acids can
be formed.
Respiration is used with the aerobic and facultative
anaerobic organisms. Respiration includes glycolysis,
Krebs tricarboxylic-acid cycle, and the electron trans-
port chain coupled with oxidative phosphorylation.
These pathways combine to produce ATP.
Obligate intracellular organisms are not capable
of the metabolic pathways for ATP synthesis and thus
must steal ATP from their host. These bacteria live in
their host cell and cannot survive without the host.
Further metabolic differences (such as sugar sources
used, end products formed, and the specific need for cer-
tain nutrients) figure in classifying bacteria and will be
discussed in the chapters covering specific organisms.
OBLIGATE
AEROBES
FACULTATIVE
ANAEROBES
MICROAEROPHILIC
OBLIGATE
ANAEROBES
Gram-positive
Nocardia
(weakly acid-fast)
Bacillus cereus
Staphylococcus
Bacillus anthracis
Corynebacterium
Listeria
Actinomyces
Streptococcus
Clostridium
Gram-negative
Neisseria
Pseudomonas
Bordetella
Legionella
Brucella
Most other gram-
negative rods
Spirochetes
T
reponema
B
orrelia
L
eptospira
Campylobacter
Bacteroides
Acid-fast
Mycobacterium
Nocardia
No cell wall
Mycoplasma
CHAPTER 2. CELL STRUCTURES,
VIRULENCE FACTORS AND TOXINS
Figure 2-1
Virulent organisms are those that can cause disease.
The virulence of an organism is the degree of organism
pathogenicity. Virulence depends on the presence of cer-
tain cell structures and on bacterial exotoxins and en-
dotoxins, all of which are virulence factors.
CELL STRUCTURES AS VIRULENCE
FACTORS
Flagella
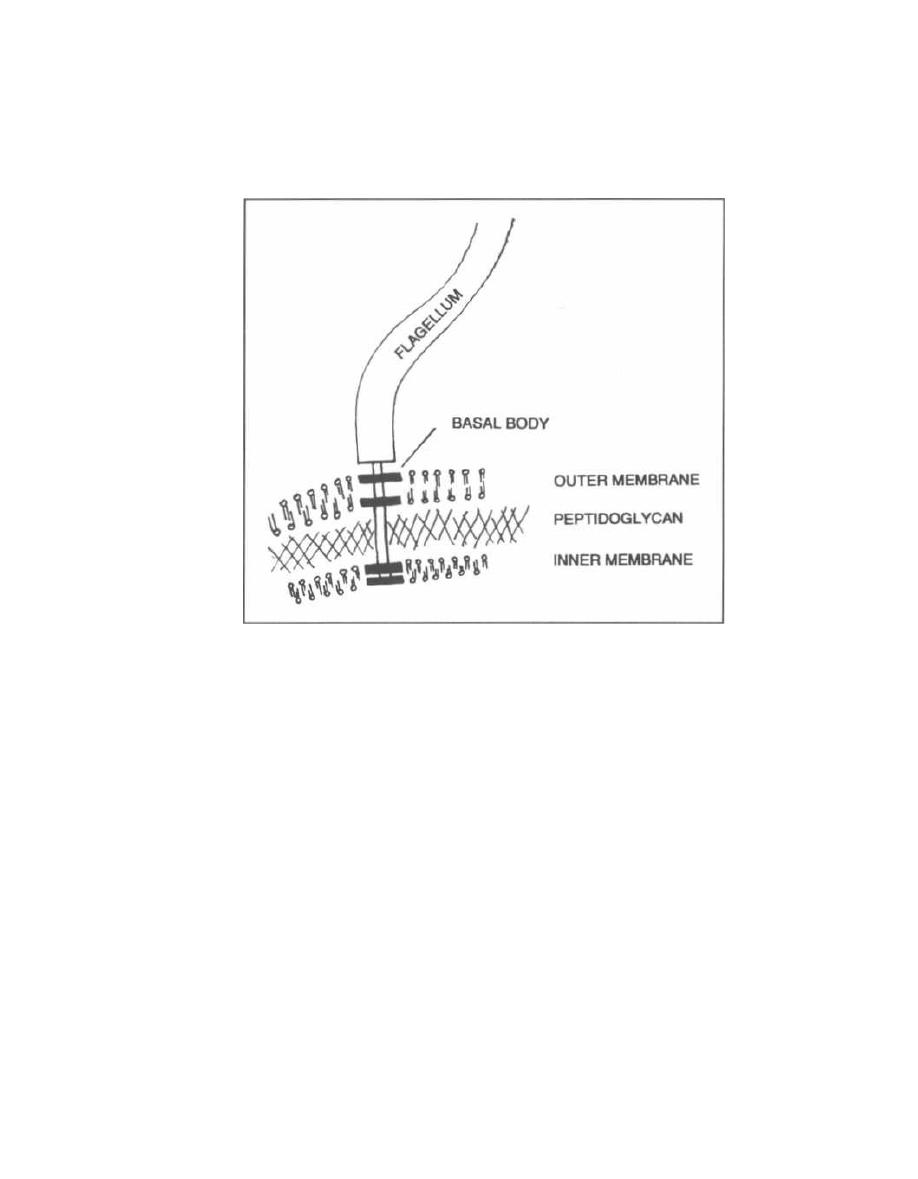
Fig. 2-1.
Flagella are protein filaments that extend
like long tails from the cell membranes of certain gram-
positive and gram-negative bacteria. These tails, which
are several times the length of the bacterial cell, move
the bacteria around. The flagellum is affixed to the bac-
teria by a basal body. The basal body spans through
the entire cell wall, binding to the inner and outer cell
membrane in gram-negative bacteria and to the inner
membrane in gram-positive bugs (the gram-positive
bacteria don't have an outer membrane). The basal body
spins around and spins the flagellum. This causes the
bacterial flagella to undulate in a coordinated manner
to move the bacteria toward a chemical concentration
8
gradient o
r
away from the gradient. This movement is
called chemotaxis.
Fig. 2-2.
Bacteria can have a single polar flagellum
( polar means at one end of the cell) as is the case with
Vibrio cholera, or many peritrichous flagella (all
around the cell) as is the case with Escherichia coli and
Proteus mirabilis. Shigella does not have flagella.
Pili
Pili (also called fimbriae) are straight filaments
arising from the bacterial cell wall, making the bac-
terium look like a porcupine.
Fig. 2-3.
Pili are much shorter than flagella and do
not move. Pili can serve as adherence factors (in which
case they are called adhesins). Many bacteria possess
adhesins that are vital to their ability to cause disease.
For example, Neisseria gonorrhea has pili that allow it
to bind to cervical cells and buccal cells to cause gonor-
rhea. Escherichia coli and Campylobacter jejuni cannot
cause diarrhea without their adhesins to bind to the in-
testinal epithelium, and Bordetella pertussis uses its
adhesin to bind to ciliated respiratory cells and cause
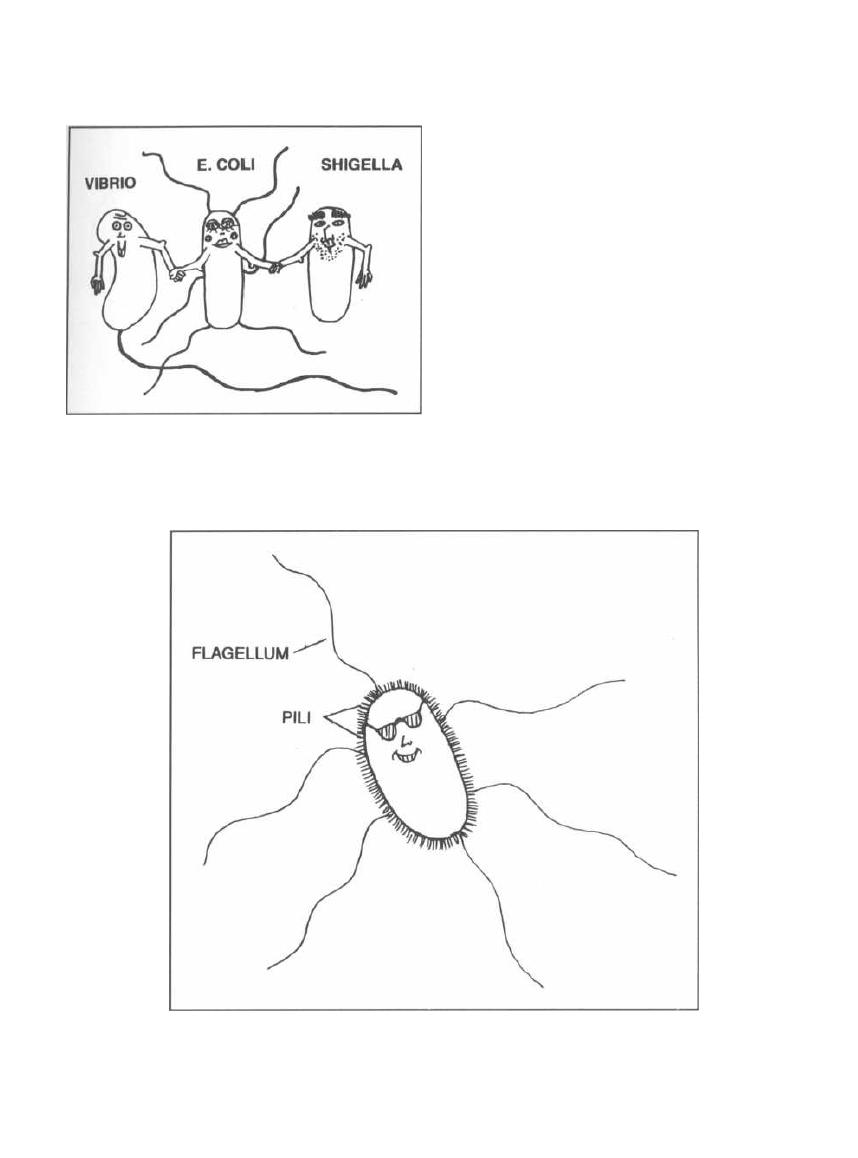
CHAPTER 2. CELL STRUCTURES, VIRULENCE FACTORS AND TOXINS
Figure 2-2
Figure 2-3
9
whooping cough. Bacteria that do not produce these pili
cannot grab hold of their victim; they lose their viru-
lence and thus cannot infect humans. There are also
special pili, discussed in the next chapter, called sex pili.
Capsules
Capsules are protective walls that surround the
cell membranes of gram-positive and gram-negative
bacteria. They are usually composed of simple sugar
residues. Bacteria secrete these sugar moieties, which
then coat their outer wall. One bacterium, Bacillus an-
thracis, is unique in that its capsule is made up of amino
acid residues.
Fig. 2-4. Capsules enable bacteria to be more viru-
lent because macrophages and neutrophils are unable
to phagocytize the encapsulated buggers. For exam-
ple, Streptococcus pneumoniae has a capsule. When
grown on media, these encapsulated bacteria appear as
smooth (S) colonies that cause rapid death when in-
jected into mice. Some Streptococcus pneumoniae do not
have capsules and appear as rough (R) colonies on agar.
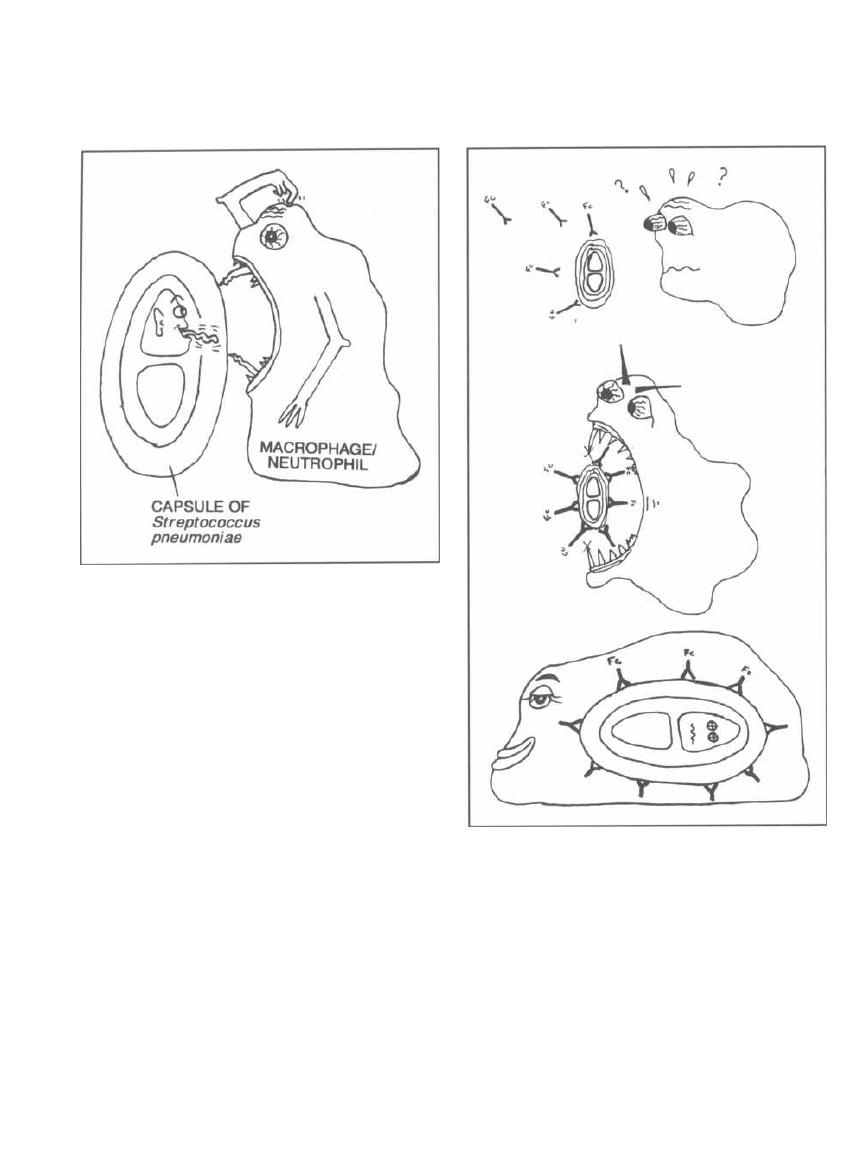
CHAPTER 2. CELL STRUCTURES, VIRULENCE FACTORS AND TOXINS
Figure 2-4
These rough colonies have lost their virulence and when
injected into mice do not cause death.
Two important tests enable doctors to visualize
capsules under the microscope and aid in identifying
bacteria:
1) India ink stain: Because this stain is not taken
up by the capsule, the capsule appears as a transparent
halo around the cell. This test is used primarily to iden-
tify the fungus
Cryptococcus.
2) Quellung reaction: The bacteria are mixed with
antibodies that bind to the capsule. When these anti-
bodies bind, the capsule swells with water, and this can
be visualized microscopically.
Antibodies directed against bacterial capsules protect
us as they help our macrophages and neutrophils bind
to and eat the encapsulated bacteria. The process of an-
tibodies binding to the capsule is called opsonization.
Fig. 2-5. Once the antibodies have bound to the
bacterial capsule
(opsonization),
the macrophage or
neutrophil can then bind to the Fc portion of the anti-
body and gobble up the bacteria. A vaccine against
Streptococcus pneumoniae
contains antigens from the
23 most common types of capsules. Immunization with
this vaccine elicits an immune response against the
capsular antigens and the production of antibodies
10
Figure 2-5
that protects the individual against future infections
by this organism.
Endospores
Endospores are formed by only 2 genera of bacteria,
both of which are gram-positive: the aerobic
Bacillus
and the anaerobic
Clostridium.
Fig. 2-6. Endospores are metabolically dormant
forms of bacteria that are resistant to heat (boiling),
cold, drying and chemical agents. They have a multi-
layered protective coat consisting of:
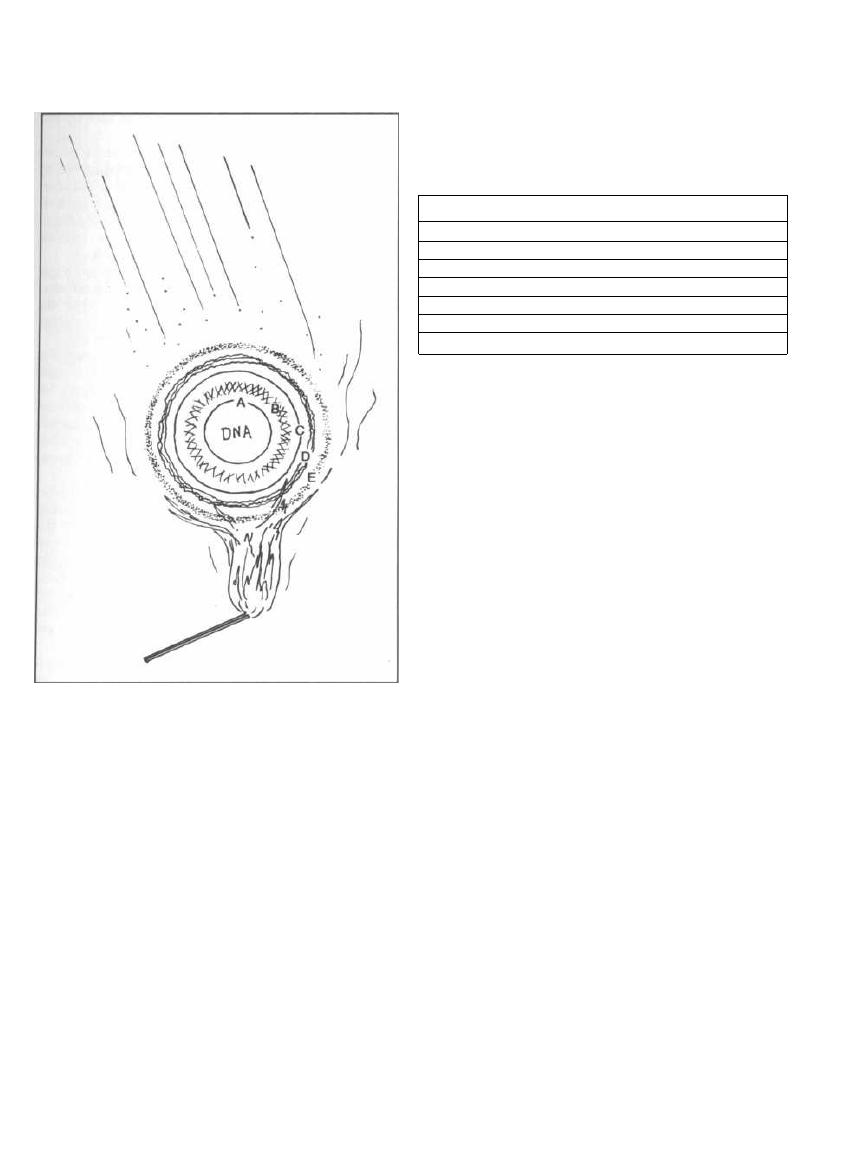
Figure 2-6
A) A cell membrane
B) A thick peptidoglycan mesh
C) Another cell membrane
D) A wall of keratin-like protein
E) An outer layer called the exosporium
Spores form when there is a shortage of needed nu-
trients and can lie dormant for years. Surgical instru-
ments are heated in an autoclave, which uses steam
under pressure, to 121°C for 15 minutes, in order to en-
sure the destruction of
Clostridium
and
Bacillus
spores.
When the spore is exposed to a favorable nutrient or en-
vironment, it becomes active again.
Facultative Intracellular Organisms
Many bacteria are phagocytosed by the host's
macrophages and neutrophils yet survive within these
white blood cells unharmed!!! These bacteria inhibit
CHAPTER 2. CELL STRUCTURES, VIRULENCE FACTORS AND TOXINS
1 1
phagosome-lysosome fusion, thus escaping the host's
deadly hydrogen peroxide and superoxide radicals. In-
side the cells these bacteria are safe from antibodies and
other immune defenses.
Figure 2-7
Fig. 2-7. Facultative intracellular organisms.
TOXINS
Exotoxins
Exotoxins are proteins that are released by both
gram-positive and gram-negative bacteria. They may
cause many disease manifestations. Essentially, exo-
toxins are released by all the major gram-positive
genera except for
Listeria monocytogenes,
which pro-
duces endotoxin. Gram-negative bacteria such as
Vibrio
cholera, Escherichia coli,
and others can also excrete ex-
otoxins. Severe diseases caused by bacterial exotoxins
include anthrax (Saddam Hussein's threatened germ
warfare agent), botulism, tetanus, and cholera.
Neurotoxins are exotoxins that act on the nerves or
motor endplates to cause paralysis. Tetanus toxin and
botulinum toxin are examples.
Enterotoxins are exotoxins that act on the gas-
trointestinal (GI) tract to cause diarrhea. Enterotoxins
inhibit NaCl resorption, activate NaCl secretion, or kill
intestinal epithelial cells. The common end result is
the osmotic pull of fluid into the intestine, which
causes diarrhea. The enterotoxins cause 2 disease
manife- stations:
1) Infectious diarrhea: Bacteria colonize and bind
to the GI tract, continuously releasing their enterotox-
ins locally. The diarrhea will continue until the bacteria
are destroyed by the immune system or antibiotics (or
the patient dies secondary to dehydration). Examples:
Vibrio cholera, Escherichia coli, Campylobacter jejuni,
and
Shigella dysenteriae.
2) Food poisoning: Bacteria grow in food and re-
lease enterotoxin in the food. The enterotoxin is in-
gested resulting in diarrhea and vomiting for less than
24 hours. Examples:
Bacillus cereus
and
Staphylococcus
aureus.
FACULTATIVE INTRACELLULAR ORGANISMS
1. Listeria monocytogenes
2. Salmonella typhi
3. Yersinia
4. Francisella tularensis
5. Brucella
6. Legionella
7. Mycobacterium

CHAPTER 2. CELL STRUCTURES, VIRULENCE FACTORS AND TOXINS
Pyrogenic exotoxins stimulate the release of cy-
tokines and can cause rash, fever, and toxic shock syn-
drome (see page 33). Examples: Staphylococcus aureus
and Streptococcus pyogenes.
Tissue invasive exotoxins allow bacteria to de-
stroy and tunnel through tissues. These include en-
zymes that destroy DNA, collagen, fibrin, NAD, red
blood cells, and white blood cells.
Miscellaneous exotoxins, which are the principle
virulence factors for many bacteria, can cause disease
unique to the individual bacterium. Often the exact role
of the exotoxin is poorly understood.
Fig. 2-S.
This chart gives many of the important exo-
toxins and compares their mechanisms of action. Glance
over the chart now and return to it as you study indi-
vidual bacteria.
Fig. 2-9.
Exotoxin subunits in Bacillus anthracis,
Clostridium botulinum, Clostridium tetani, Corynebac-
terium diphtheriae, and Vibrio cholera. Their exotoxins
are all composed of 2 polypeptide subunits bound to-
gether by disulfide bridges. One of these subunits
(called B for binding or H for holding on) binds to the
target cell. The other subunit (called A for action or L
for laser) then enters the cell and exerts the toxic effect.
Picture these subunits as a key (B and H) and a gun (A
and L) bound together by disulfide bonds. The key opens
the cell, and then the gun does its damage.
Endotoxins
Remember from the last chapter that endotoxin is
lipid A, which is a piece of the outer membrane
lipopolysaccharide (LPS) of gram-negative bacteria (see
Fig. 1-6). Lipid A/endotoxin is very toxic and is released
when the bacterial cell undergoes lysis (destruction).
Endotoxin is also shed in steady amounts from living
bacteria. Sometimes, treating a patient who has a gram-
negative infection with antibiotics can worsen the pa-
tient's condition because all the bacteria are lysed,
releasing large quantities of endotoxin. Endotoxin dif-
fers from exotoxin in that it is not a protein ex-
creted from cells, but rather is a normal part of
the outer membrane that sort of sheds off, espe-
cially during lysis. Endotoxin is ONLY present in
gram-negative bacteria with one exception: Listeria
monocytogenes, a gram-positive bacteria, has endotoxin.
Septic Shock
Septic shock (endotoxic shock) is a common and
deadly response to both gram-negative and gram-
positive infection. In fact, septic shock is the number
one cause of death in intensive care units and the 13th
most common cause of death in the U.S. (Parrillo, 1990).
1 2
To better understand septic shock, let us back up and
review some terms.
Bacteremia: This is simply bacteria in the blood-
stream. Bacteria can be detected by isolating the of-
fending critters in blood cultures. Bacteremia can occur
silently and without symptoms. Brushing your teeth re-
sults in transient bacteremia with few systemic conse-
quences. Bacteremia can also trigger the immune
system, resulting in sepsis and possibly death.
Sepsis: Sepsis refers to bacteremia that causes a sys-
temic immune response to the infection. This response
can include high or low temperature, elevation of the
white blood cell count, and fast heart rate or breathing
rate. Septic patients are described as "looking sick."
Septic shock: Sepsis that results in dangerous drops
i n blood pressure and organ dysfunction is called septic
shock. It is also referred to as endotoxic shock be-
cause endotoxin often triggers the immune response
that results in sepsis and shock. Since gram-positive
bacteria and fungi can also trigger this adverse immune
response, the term septic shock is more appropriate
and inclusive.
The chain of events that lead to sepsis and often
death begins with a localized site of infection of gram-
negative or gram-positive bacteria or fungi. From this
site or from the blood (bacteremia), the organisms re-
lease structural components (such as endotoxin and/or
exotoxin) that circulate in the bloodstream and stimu-
late immune cells such as macrophages and neu-
trophils. These cells, in response to the stimulus,
release a host of proteins that are referred to as en-
dogenous mediators of sepsis.
The most famous endogenous mediator of sepsis is
tumor necrosis factor (TNF). TNF is also called
cachectin because it is released from tumors, produc-
ing a wasting (weight loss) syndrome, called cachexia,
in cancer patients. Injecting TNF into experimental
animals produces hypotension and death (septic shock).
In sepsis, TNF triggers the release of the cytokine
interleukin-1 from macrophages and endothelial cells,
which in turn triggers the release of other cytokines
and prostaglandins. This churning maelstrom of medi-
ators at first defends the body against the offending
microorganisms, but ultimately turns against the body.
The mediators act on the blood vessels and organs
to produce vasodilatation, hypotension, and organ sys-
tem dysfunction.
The mortality rate for septic shock is high: up to 40%
of patients will die, even with intensive care and an-
tibiotic therapy. For every organ system that fails the
mortality rises. Usually two organs are involved (vas-
cular system with hypotension and lungs with hypoxia)
and the mortality rate is about 40%. For each addi-
tional organ failure (renal failure, etc.) add 15-20%
mortality!
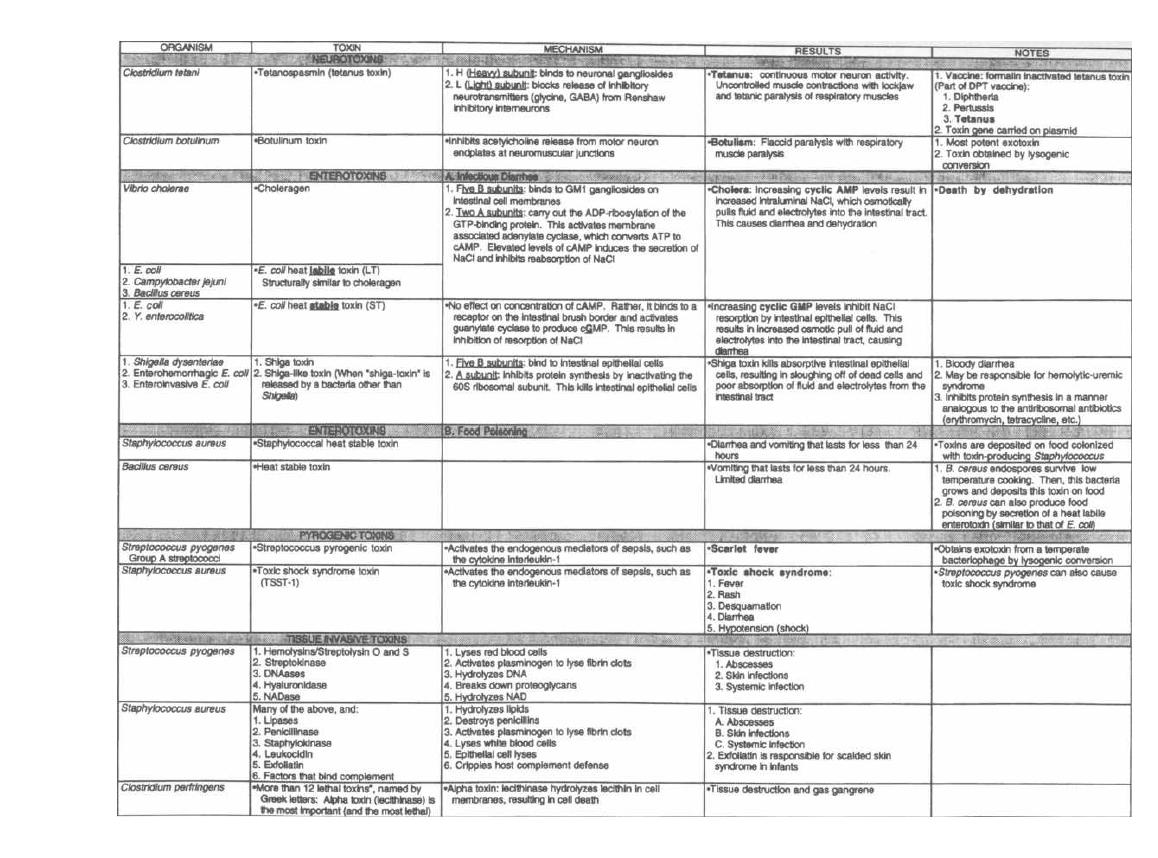
Figure 2-8
EXOTOXINS
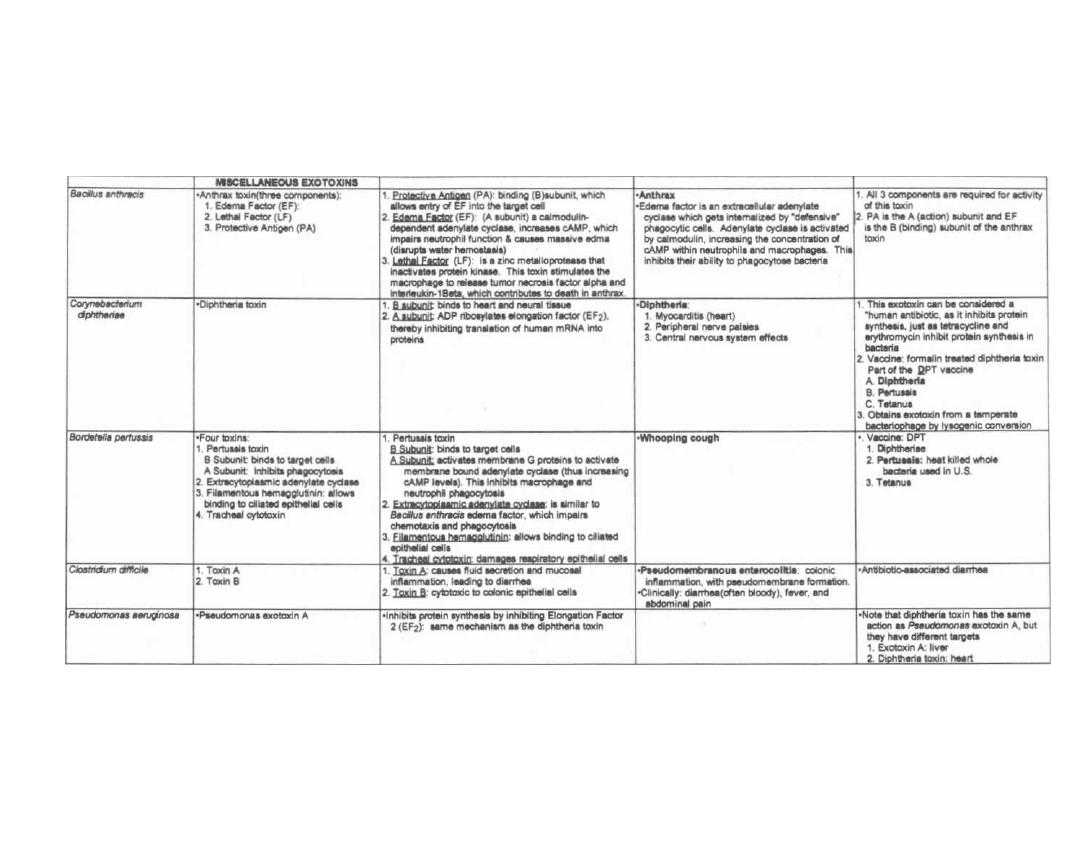
Figure 2-8 (continued)
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
©MedMaster
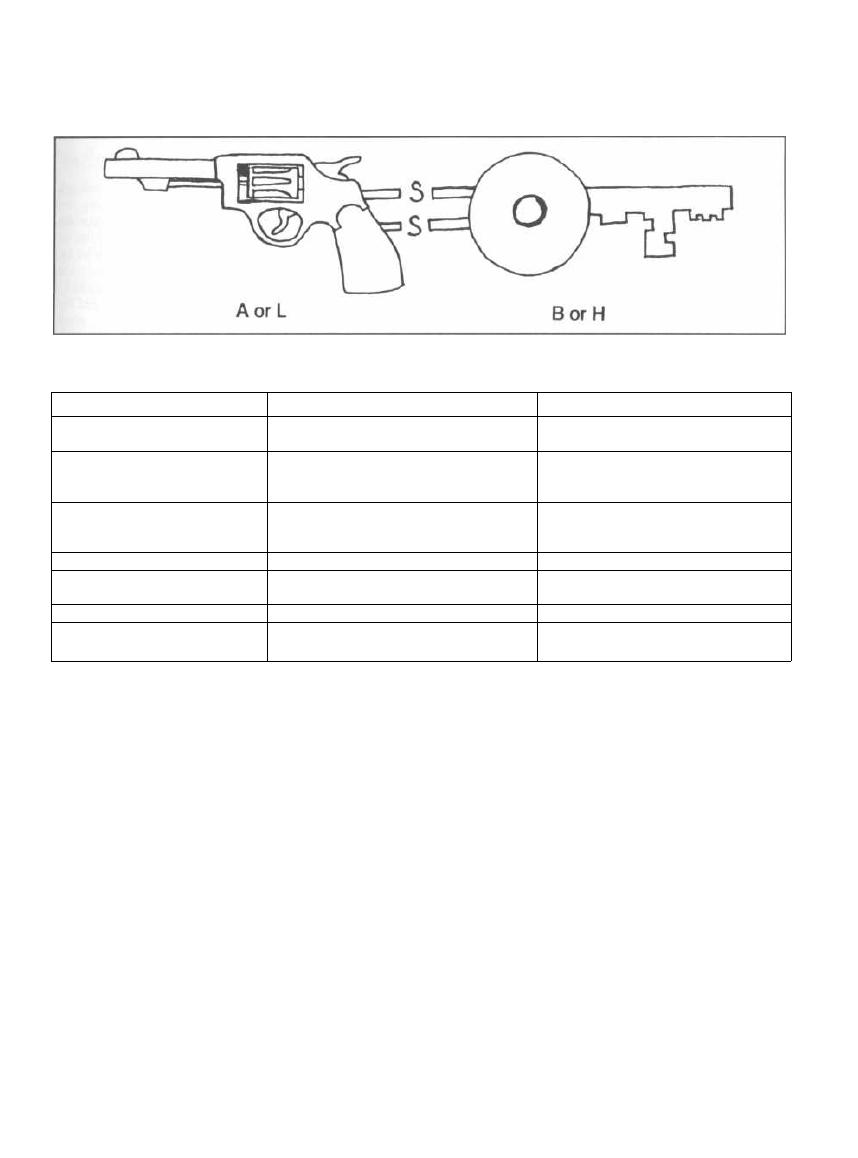
Figure 2-9
Figure 2-10
EFFECTS OF SEPTIC SHOCK
Treatment
The most important principle of treatment is to find
the site of infection and the bug responsible and eradi-
cate it! The lung is the most common site (pneumonia)
followed by the abdomen and urinary tract. In one-third
of cases a site of infection is not identified. Antibiotic
therapy is critical with a 10 to 15 fold increased mortal-
ity when antibiotics are delayed. Even while working up
the site of infection you should start broad coverage an-
tibiotics (called empiric therapy). In other words, as soon
as the patient looks sick, start blasting your shotgun at
all potential targets. Fire early and hit everything.
Blood pressure must be supported with fluids and
drugs (dopamine and norepinephrine are commonly
used) and oxygenation maintained (intubation and me-
chanical ventilation is often required).
In the last decades, efforts to block the inflammatory
cascade with monoclonal antibodies against endotoxin,
tumor necrosis factor, and interleukin-1, anti-inflam-
matory agents such as ibuprofen and steroids, and a
CHAPTER 2. CELL STRUCTURES, VIRULENCE FACTORS AND TOXINS
15
host of other investigational agents (tumor necrosis
factor soluble receptor, nitric oxide antagonists, and
antioxidant compounds), have met with disappointing
results. Most of these treatments have failed to reduce
mortality in clinical trials.
Fig. 2-10.
The end organ effects of septic shock.
References
Parrillo JE. Pathogenic mechanisms of septic shock. N Engl J
Med
1993;328:1471-1477.
Parrillo JE, moderator. Septic shock in humans: advances in
the understanding of pathogenesis, cardiovascular dys-
function, and therapy. Ann Intern Med
1990;113:227-42.
Recommended Review Article:
Wheeler, AP, Bernard GR. Treating patients with septic
shock. N Engl J Med
1999;340:207-214.
ORGAN SYSTEM
EFFECT ON ORGAN SYSTEM
DAMAGING EFFECT ON BODY
Vascular system
Vasodilation
1. Decreased blood pressure
2. Organ hypoperfusion
Heart
Myocardial depression
1. Decreased cardiac output
2. Decreased blood pressure
3. Organ hypoperfusion
Kidneys
Acute renal failure
1. Decreased urine output
2. Volume overload
3. Accumulation of toxins
Lungs
Adult respiratory distress syndrome
H
ypoxia
Liver
Hepatic failure
1. Accumulation of metabolic toxins
2. Hepatic encephalopathy
Brain
Encephalopathy
A
lteration in mental status
Coagulation system
Disseminated intravascular coagulation 1. Clotting
2. Bleeding

CHAPTER 3. BACTERIAL SEX GENETICS
The bacterial chromosome is a double-stranded DNA
molecule that is closed in a giant loop. Because there is
only one copy of this molecule per cell, bacteria exist in
a haploid state. Bacteria do not have nuclear mem-
branes surrounding their DNA.
This chapter does not attempt to cover all the details
of bacterial genetics, such as replication, transcription,
and translation. These topics are covered extensively in
genetics courses. Instead, this chapter covers the mech-
anisms of bacterial exchange of genetic information.
You see, procaryotes have it rough as they do not engage
i n sexual union with other bacteria. They undergo gene
replication, forming an exact copy of their genome, and
then split in two, taking a copy with each half (binary
fission). The cells of higher organisms (eucaryotes) con-
tribute a set of gametes from each parent and thus en-
sure genetic diversity. So how do the sexless creatures
undergo the genetic change so necessary for survival?
One mechanism is simple mutation. However, it is
rare for a single point mutation to change an organism
i n a helpful manner. Point mutations usually result in
nonsense or missense (does this make sense?). There
are 4 ways in which bacteria are able to exchange ge-
netic fragments: 1) transformation, 2) transduction, 3)
conjugation (so much for celibacy), and 4) transposon
insertions
.
CHANGE = SURVIVAL
The exchange of genetic material allows for the shar-
i ng of genes that code for proteins, such as those that
provide antibiotic resistance, exotoxins, enzymes, and
other virulence factors (pili, flagella, and capsules).
Scientists can take advantage of these exchange mech-
anisms for genetic engineering and chromosomal map-
ping. Read on ... but only if you are over 21 years old.
TRANSFORMATION
Naked DNA fragments from one bacterium, released
during cell lysis, bind to the cell wall of another bac-
terium. The recipient bacterium must be competent,
which means that it has structures on its cell wall that
can bind the DNA and take it up intracellularly. Recip-
i ent competent bacteria are usually of the same species
as the donor. The DNA that has been brought in can
then incorporate itself into the recipient's genome if
there is enough homology between strands (another
reason why this transfer can only occur between closely
related bacteria).
The famous example of this type of exchange is the ex-
periment conducted by Frederick Griffith in 1928. He
used the Streptococcus pneumoniae bacteria, which are
classified into many different types based on differences
16
in their cellular capsule. Griffith used smooth encapsu-
l ated pneumococci, which cause violent infection and
death in mice, and rough nonencapsulated pneumo-
cocci, which do not kill mice. You can think of the en-
capsulated pneumococci as smooth hit men who kill
mice, and the rough nonencapsulated pneumococci as
only acting rough (they are pushovers and can't kill a
flea). Griffith heat-killed the smooth encapsulated bad
guys and injected them, along with the live rough
nonencapsulated pushovers, into mice. Lo and behold,
the mice died, and when he cultured out bacteria from
the blood, he could only find live smooth encapsulated
pneumococci. The gene encoding the capsule had been
released from the heat-killed bacteria and became in-
corporated into the living rough nonencapsulated bac-
teria. The rough bacteria were thus transformed into
virulent encapsulated smooth bacteria.
Scientists now use this method extensively for in-
serting recombinant DNA and for mapping genes on
chromosomes. It can be used in mapping because the
frequency of transformation leading to two traits being
transferred is relative to their distance apart on the
genome. The closer they are to each other, the more
likely that they will be transferred together.
TRANSDUCTION
Transduction occurs when a virus that infects bacte-
ria, called a bacteriophage, carries a piece of bacter-
ial DNA from one bacterium to another. To understand
this topic, let us digress for a moment and talk about
bacteriophages.
Fig. 3-1.
Bacteriophages resemble most viruses in
having a protein coat called a capsid that surrounds a
molecule of DNA or RNA. They look almost like spiders
with long skinny necks.
The phage will bind by its tail fibers to specific recep-
tors on the bacterial cell surface. This is called adsorp-
tion. The phage then undergoes penetration. Much
like a spider squatting down and sinking in its stinger,
the phage pushes the long hollow tube under its neck
sheath through the bacterial cell wall and cytoplasmic
membrane. DNA in the head is injected through the
tube into the bacterium.
Fig. 3-2.
Following adsorption and penetration, the
i njected DNA takes over the host bacteria's RNA poly-
merase for the transcription of phage DNA to mesenger
RNA (mRNA). New capsids, DNA, and enzymes are
formed, and the bacterial cell fills with new phages. At
some point the cell can hold no more particles and lyses,
releasing the phages.
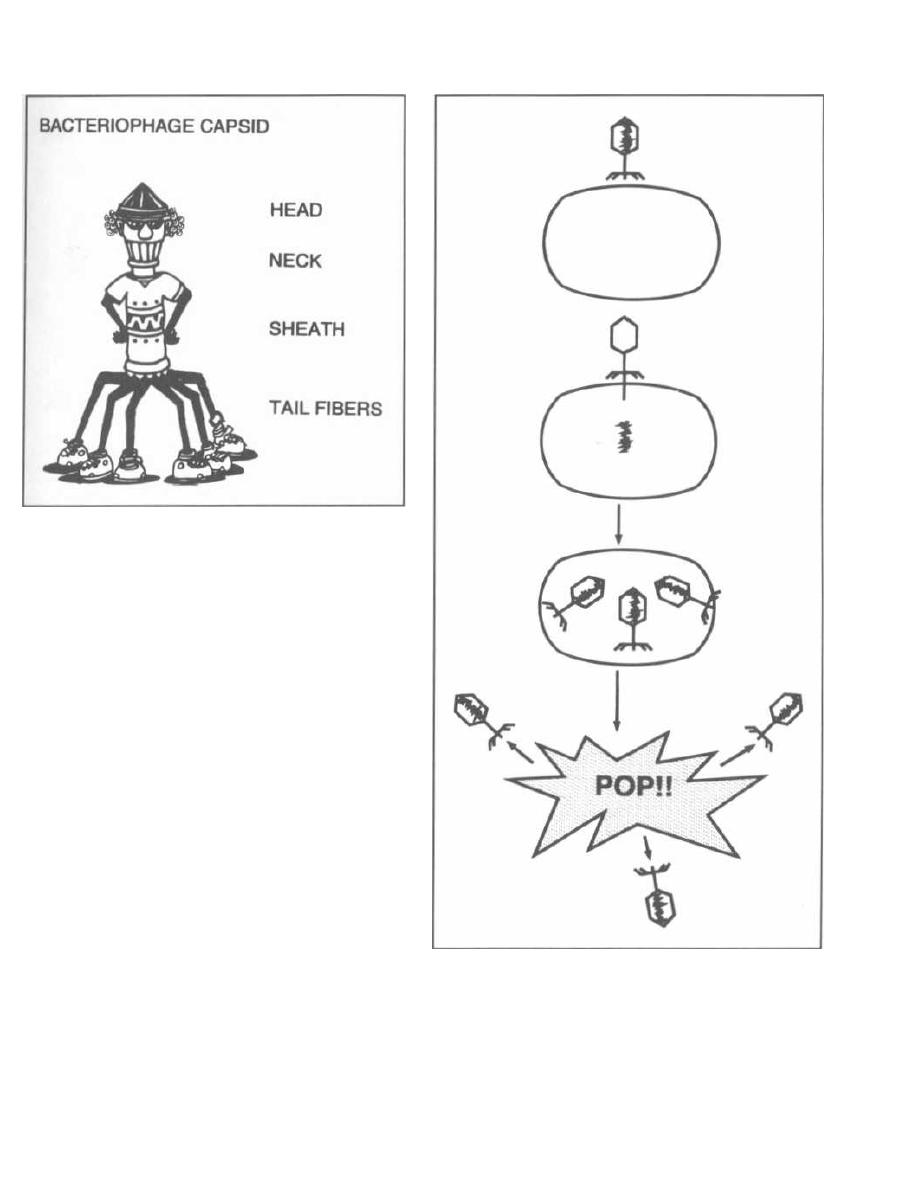
Figure 3-1
To make things more complicated, there are two types
of phages, virulent phages and temperate phages.
Virulent phages behave as shown in Fig. 3-2, infecting
the bacteria, reproducing, and then lysing and killing
the bacteria. On the other hand, temperate phages have
a good temperament and do not immediately lyse the
bacteria they infect. The temperate phage undergoes ad-
sorption and penetration like the virulent phage but
then, rather than undergoing transcription, its DNA be-
comes incorporated into the bacterial chromosome. The
DNA then waits for a command to activate.
Fig. 3-3.
The integrated temperate phage genome is
called a prophage. Bacteria that have a prophage
integrated into their chromosome are called lysogenic
because at some time the repressed prophage can be-
come activated. Once activated, the prophage initiates
the production of new phages, beginning a cycle that
ends with bacterial cell lysis. So temperate phages,
although of good temperament, are like little genetic
time bombs.
Lysogenic immunity is the term used to describe
the ability of an integrated bacteriophage (prophage) to
block a subsequent infection by a similar phage. The
first temperate phage to infect a bacteria produces a re-
pressor protein. This "survival of the fittest" adaptation
terium to another. This process is called transduction.
ensures that the first temperate phage is the bacteria's
Just as there are two types of phages, there are two
sole occupant.
types of transduction. Virulent phages are involved in
Now that we understand bacteriophages, let's discuss
generalized transduction and temperate phages in
how these phages can carry bacterial DNA from one bac-
specialized transduction.
CHAPTER 3. BACTERIAL GENETICS
1 7
Figure 3-2
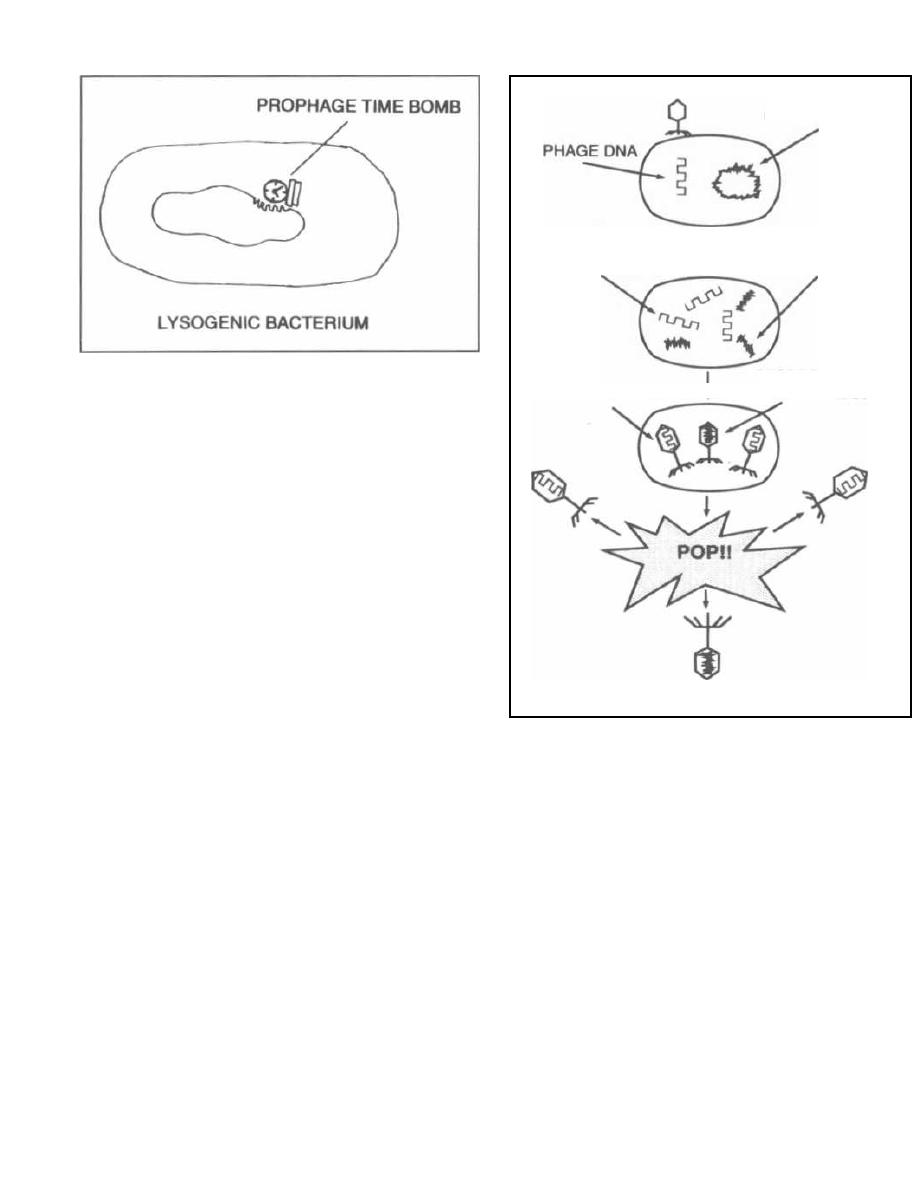
Figure 3-3
Generalized Transduction
Generalized transduction occurs as follows. After
phage penetration into a host bacterium, the phage
DNA is transcribed, replicated, and translated into
capsids and enzymes. At this same time the bacterial
DNA is repressed and eventually destroyed. Some-
times pieces of the bacterial DNA are left intact. If
these pieces are the same size as the phage DNA, they
can accidentally be packed into the phage capsid head.
Following lysis of the cell and release of the phages, the
one phage with bacterial DNA in its head can then in-
fect another bacterium. It will inject the piece of bacte-
rial DNA that it is "accidentally" carrying. If there is
some homology between the newly injected strand and
the recipient bacterial genome, the piece may become
incorporated. The gene on that piece could encode a
protein that the recipient did not originally have, such
as a protein that inactivates an antibiotic. In general-
ized transduction, the bacteriophage is only carrying
bacterial DNA, so the recipient cell will survive (since
no viral genes that encode for replication and lysis are
present). This type of genetic transfer is more effective
than transformation because the transferred DNA
piece is protected from destruction during transfer by
the phage capsid that holds it.
Fig. 3-4. Generalized transduction
A) Adsorption and penetration occur. The viral DNA
is drawn as a thin line, and the bacterial circular DNA
is drawn as a thick circle.
B) Destruction of the bacterial DNA leaves some in-
tact (thick) pieces. The phage DNA has undergone
replication.
C) Capsids are translated and packed. The middle
one has been packed with a bacterial DNA fragment.
D) Cell lysis occurs, liberating phages including the
phage with bacterial DNA.
CHAPTER 3. BACTERIAL GENETICS
18
REPLICATED
PHAGE DNA
MISPACKAGED
PHAGE DNA
BACTERIAL DNA
PHAGE WITH BACTERIAL DNA
BACTERIAL DNA
DISRUPTED
BACTERIAL
DNA
Figure 3-4
Specialized Transduction
Specialized transduction occurs with temperate
phages. Remember that the temperate phage pene-
trates, and then its DNA becomes incorporated into the
bacterial chromosome. It is then called a prophage, and
the bacterium is now lysogenic (Fig. 3-3). Normally the
prophage just waits doing nothing, but it can eventually
become active. If it becomes active, the prophage DNA
is spliced out of the bacterial chromosome and is then
replicated, translated, and packaged into a capsid.
Sometimes there is an error in splicing, and a piece of
bacterial DNA that lies at one side of the prophage will
be cut, replicated, and packaged with the phage DNA.
This may result in a transfer of that piece of bacterial
DNA to another bacteria.
Fig. 3-5. Specialized transduction occurs with
phage lambda in Escherichia coli. The site of insertion
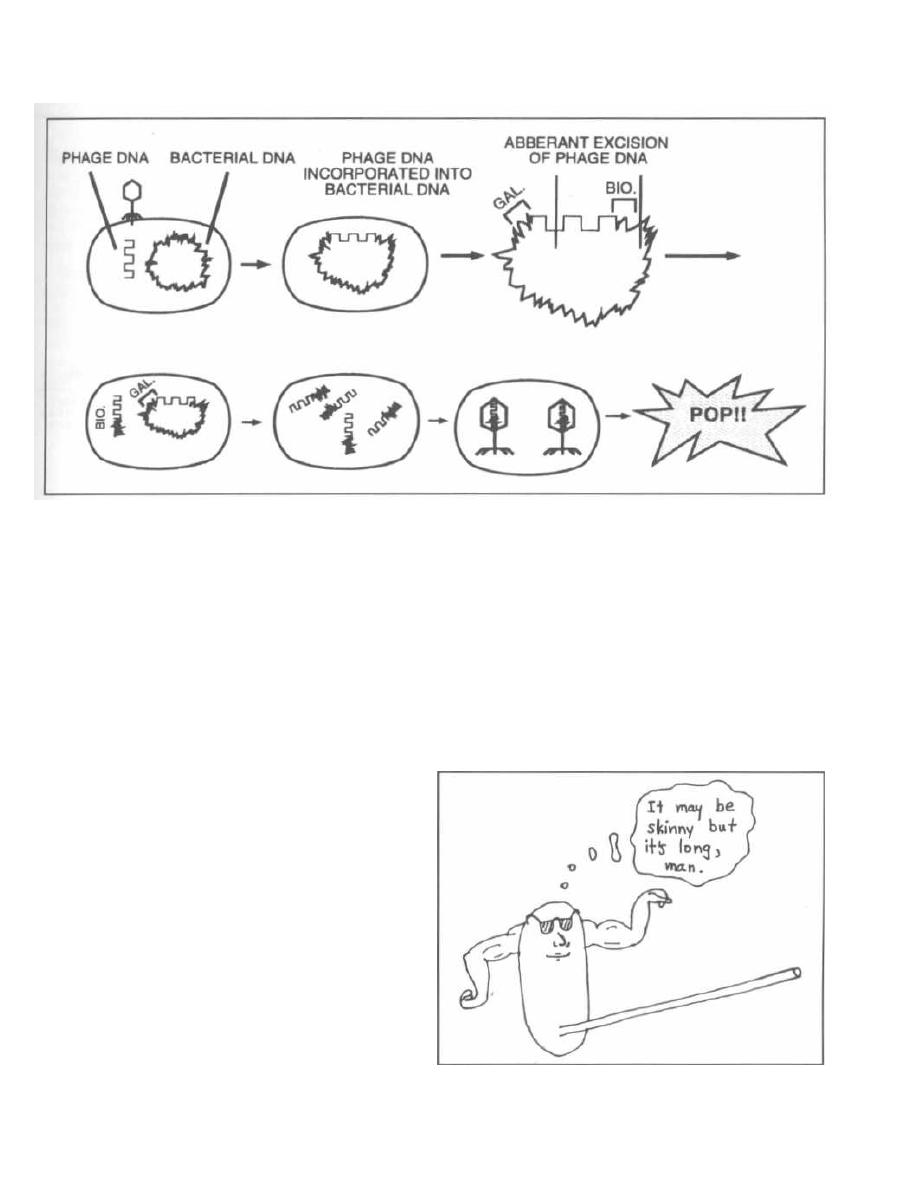
Figure 3-5
of the lambda prophage lies between the
Escherichia
coli
gene for biotin synthesis and galactose synthesis. If
a splicing error occurs, the biotin (BIO) gene or the
galactose (GAL) gene (but not both, as the piece of DNA
spliced is of a set length) will be carried with the phage
DNA and packaged. Thus the gene for biotin synthesis
can now be transferred to another bacteria that does not
have that capability. You will frequently hear about this
form of gene acquisition; it is called lysogenic conver-
sion. For example, the gene for
Corynebacterium diph-
theria's
exotoxin is obtained by lysogenic conversion.
CONJUGATION
Conjugation is bacterial sex at its best: hot and heavy!
In conjugation DNA is transferred directly by cell-to-cell
contact, resulting in an extremely efficient exchange of
genetic information. The exchange can occur between
unrelated bacteria and is the major mechanism for
transfer of antibiotic resistance.
For conjugation to occur, one bacterium must have a
self-transmissible plasmid, also called an F plasmid
(for fertility, not the other word!). Plasmids are circular
double-stranded DNA molecules that lie outside the
chromosome and can carry many genes, including those
for drug resistance. F plasmids encode the enzymes and
proteins necessary to carry out the process of conjuga-
tion. Bacteria that carry F plasmids are called F(+)
cells. In conjugation, an F(+) donor cell will pass its F
plasmid to an F( -) recipient cell, thus making the re-
cipient F(+).
CHAPTER 3. BACTERIAL GENETICS
1 9
Fig. 3-6.
The self-transmissible plasmid (F plasmid)
has a gene that encodes enzymes and proteins that form
the sex penis, that is, sex pilus.
This long protein structure protrudes from the cell
surface of the donor F(+) bacterium and binds to and
penetrates the cell membrane of the recipient bacterium
(this is finally getting juicy!). Now that a conjugal bridge
has formed, a nuclease breaks off one strand of the F
plasmid DNA, and this single strand of DNA passes
through the sex pilus (conjugal bridge) to the recipient
bacterium.
Figure 3-6
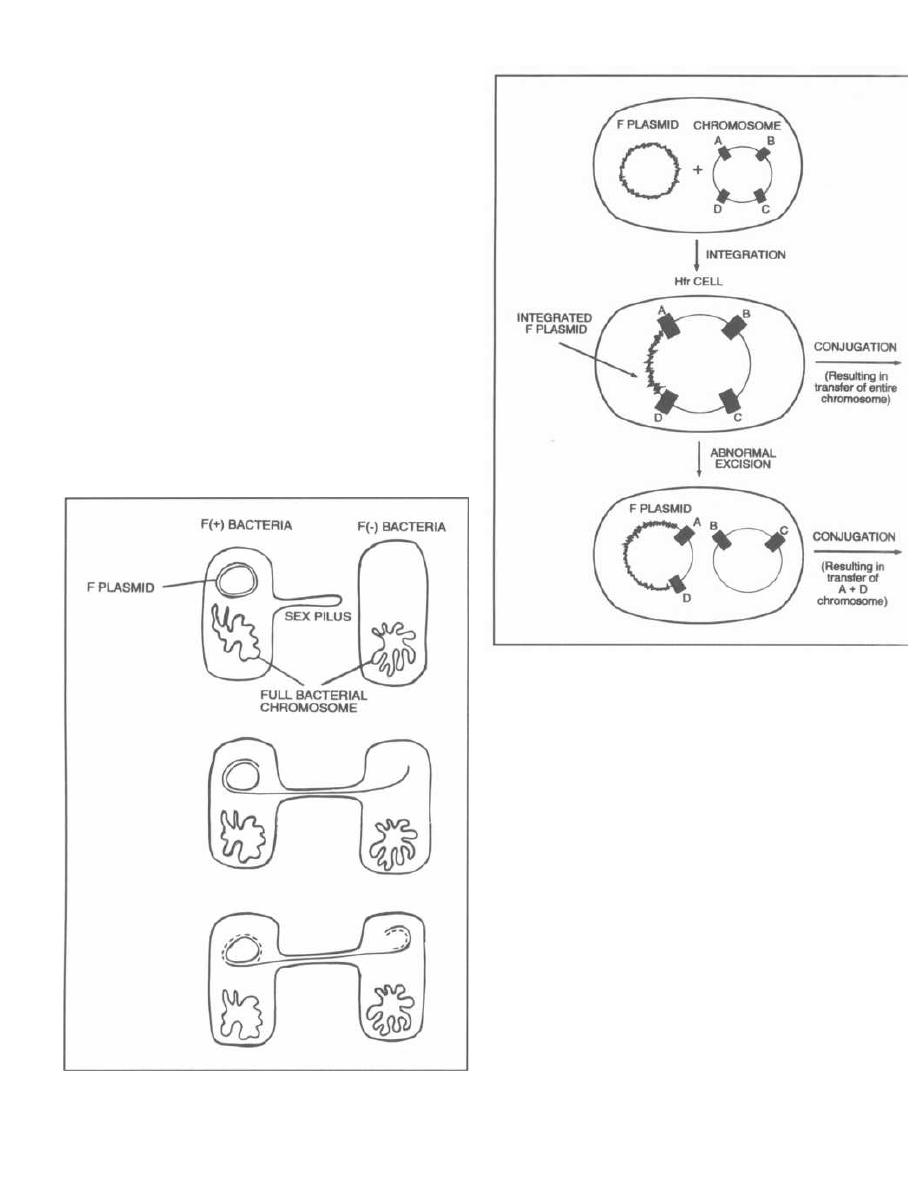
Fig. 3-7.
As one DNA strand is passed through the
conjugal bridge, the remaining strand is paired with
new nucleotide bases (dotted line). The same thing hap-
pens to the strand that passes to the other cell. At the
end of the sexual union, the conjugal bridge breaks
down and both bacteria have double-stranded circular F
plasmids. The recipient F(-) cell is now F(+).
Fig. 3-8.
Rarely, the extra-chromosomal F plasmid
becomes integrated in the neighboring bacterial chro-
mosome much in the same way as a temperate bacte-
riophage does. The bacterial cell is then called a Hfr
cell (High frequency of chromosomal recombinants).
This integration can result in two unique mechanisms
of DNA transfer:
1) The F plasmid that is now together with the entire
bacterial circular DNA undergoes normal conjugation
Figure 3-7
CHAPTER 3. BACTERIAL GENETICS
20
Figure 3-8
with an F( -) cell. The entire bacterial chromosome (in-
cluding the integrated F plasmid) will transfer from the
Hfr cell to the recipient cell.
2) The integrated F plasmid in the Hfr cell may be
excised at a different site from that of integration. This
can result in an F plasmid that now also contains a seg-
ment of chromosomal DNA. These plasmids are called
F' (F
prime) plasmids. This F' conjugation is analo-
gous to specialized transduction because in both situa-
tions a nearby segment of chromosomal DNA is picked
up "accidentally" and can be transferred to other bacte-
rial cells.
Some plasmids are non-self-transmissible plasmids.
These plasmids do not have the genes necessary for di-
recting conjugation. They do replicate within their host
bacterium, however, and continue to be passed on as the
bacteria divide in binary fission.
Plasmids are tremendously important medically.
Certain plasmids encode enzymes that degrade antibi-
otics (penicillinase), or generate virulence factors (such
as fimbriae and exotoxins).

TRANSPOSONS
Fig. 3.9. Transposons are mobile genetic elements.
You can visualize them as DNA pieces with legs.
These pieces of DNA can insert themselves into a donor
chromosome without having DNA homology. They
can carry genes for antibiotic resistance and viru-
lence factors.
Transposons insert into the DNA of phages, plas-
mids, and bacterial chromosomes. They do not repli-
cate independently but are copied during their host's
DNA transcription. When transposons leave the DNA
they are incorporated in, there is frequently aberrant
excision and the transposon can carry new DNA away
drug resistance can move to the plasmids of different
to another site. The importance of transposons clivi-
bacterial genera, resulting in the rapid spread of resis-
cally is that a transposon gene that confers a particular
taut strains.
CHAPTER 3. BACTERIAL GENETICS
Figure 3-9

Tests for Strep and Staph
Streptococci and staphylococci are both gram-positive
spheres (cocci) and are responsible for a wide variety of
clinical diseases. It is often necessary to differentiate
between these two organisms to prescribe the appropri-
ate antibiotic. The first way to differentiate them is to
examine their appearance on a Gram stain. Streptococci
line up one after the other like a strip of button candy,
while staphylococci appear as a cluster that can be vi-
sualized as a cluster of hospital staff members posing
for a group shot (Fig. 4-1).
Fig. 4-1. A second method to differentiate streptococci
from staphylococci involves the enzyme catalase. A
quick look at our staff (Staph) picture reveals that a
CAT has joined them, so the staff picture is CAT(alase)
positive. That is, staphylococci possess the enzyme
catalase, whereas streptococci do not. Staphylococci
are thus referred to as catalase positive while strepto-
cocci are catalase negative. Catalase converts H2O2
(hydrogen peroxide, which is used by macrophages
and neutrophils) into 1120 and 0
2
. To test for catalase,
a wire loop is rubbed across a colony of gram-positive
cocci and mixed on a slide with 11202. If bubbles appear,
the enzyme catalase must be present, and so staphylo-
cocci are present. (See Fig. 5-2).
Figure 4-1
GRAM-POSITIVE BACTERIA
CHAPTER 4. STREPTOCOCCI
22
Streptoccal Classification
Certain species of streptococci can either completely
or partially hemolyze red blood cells (RBCs). The strep-
tococci are divided into three groups based on their spe-
cific hemolytic ability. The streptococci are incubated
overnight on a blood agar plate. Beta-hemolytic strep-
tococci completely lyse the RBCs, leaving a clear zone of
hemolysis around the colony. Alpha-hemolytic strep-
tococci only partially lyse the RBCs, leaving a greenish
discoloration of the culture medium surrounding the
colony. This discolored area contains unlysed RBCs and
a green-colored metabolite of hemoglobin. Gamma-
hemolytic streptococci are unable to hemolyze the
RBCs, and therefore we should really not use the word
"hemolytic" in this situation (the term non-hemolytic
streptococci is often used to avoid confusion).
The streptococci can also be classified based on the
antigenic characteristics of the C carbohydrate (a car-
bohydrate found on the cell wall). These antigens are
called Lancefield antigens and are given letter names
(from A, B, C, D, E, through S). Historically, the
Lancefield antigens have been used as a major way
of differentiating the many streptococci. However,
there are so many different types of streptococci
that we now rely less on the Lancefield antigens
and more on a combination of tests such as the
above mentioned patterns of hemolysis, antigenic
composition (including Lancefield), biochemical re-
actions, growth characteristics, and genetic stud-
ies. Although there are more than 30 species of
streptococci, only 5 are significant human pathogens.
Three of these pathogens have Lancefield antigens:
Lancefeld group A, B and D. The other two pathogenic
species of the streptococcal genus do not have Lancefield
antigens, and are therefore just called by their species
names: One is
Streptococcus pneumoniae
and the
other is actually a big group of streptococci collec-
tively called the Viridans group streptococci.
GROUP A BETA-HEMOLYTIC
STREPTOCOCCI
(also called
Streptococcus pyogenes)
These organisms are so-named because they possess
the Lancefeld group A antigen and are beta-hemolytic
on blood agar. They are also called
Streptococcus pyo-
genes
(which means pus-producing) and cause the dis-

CHAPTER 4. STREPTOCOCCI
eases "strep throat," scarlet fever, rheumatic fever, and
post-streptococcal glomerulonephritis.
The components of the streptococcal cell wall that are
antigenic include:
1) C carbohydrate: The C carbohydrate was used by
Rebecca Lancefield to divide streptococci into groups.
Streptococcus pyogenes
has the "Lancefield Group A" type
of C carbohydrate.
2) M protein (80 types): This is a major virulence fac-
tor for the group A streptococcus. It inhibits the activation
of complement and protects the organism from phagocyto-
sis. However, it is also the weakest point in the organism's
defense, because plasma (B) cells generate antibodies
against the M protein. These antibodies bind to the M pro-
tein (opsonization), aiding in the destruction of the organ-
ism by macrophages and neutrophils.
Beta-hemolytic group A streptococci also have many en-
zymes that contribute to their pathogenicity:
1) Streptolysin O: The O stands for oxygen labile as it
is inactivated by oxygen. This enzyme destroys red and
white blood cells and is the reason for the beta-hemolytic
group A streptococci's beta-hemolytic ability. This enzyme
is also antigenic. Following pharyngeal or systemic beta-
hemolytic group A streptococcal infection, anti-strep-
tolysin O (ASO) antibodies develop. On the wards you may
order ASO titers on a patient's blood to confirm recent in-
fection.
2) Streptolysin S_: The S stands for oxygen stabile.
This is also responsible for beta-hemolysis but is not anti-
genic.
3) Pyrogenic exotoxin (also called erythrogenic
toxin): This is found in only a few strains of beta-
hemolytic group A streptococci, but when these strains in-
vade they can cause scarlet fever.
Some strains produce pyrogenic exotoxins that are su-
perantigens. The exotoxins directly superstimulate T cells
to pour out inflammatory cytokines. This causes a strepto-
coccal toxic shock syndrome (Holm, 1996). More on scarlet
fever and toxic shock syndrome later. . .
4) Other enzymes include streptokinase (activates
the proteolytic enzyme plasmin, which breaks up fibrin
blood clots), hyaluronidase, DNAases, anti-C5a pepti-
dase, and others (see Fig. 2-S).
Staphylococcus aureus
has many enzymes that are sim-
ilar to those of streptococci. You will learn about these in
the next chapter.
Beta-hemolytic group A streptococci cause 4 types of dis-
ease by local invasion and/or exotoxin release. These
include:
1) Streptococcal pharyngitis
23
2) Streptococcal skin infections
3) Scarlet fever
4) Streptococcal toxic shock syndrome
Beta-hemolytic group A streptococci can also cause 2
delayed antibody mediated diseases:
1) Rheumatic fever
2) Glomerulonephritis
Local Invasion/Exotoxin Release
1) Streptococcal pharyngitis: This is the classic
strep throat with red swollen tonsils and pharynx, a pu-
rulent exudate on the tonsils, high temperature, and
swollen lymph nodes. It usually lasts 5 days (penicillin
therapy speeds recovery).
"Mom, my throat hurts!!!"
2) Skin infections: Skin infections can range from
folliculitis (infections of the hair follicles), cellulitis (a
deep infection of the skin cells, producing red, swollen
skin which is hot to the touch), and impetigo (a vesicu-
lar, blistered, eruption, most common in children, that
becomes crusty and flaky and is frequently found
around the mouth). These skin infections can also be
caused by
Staphylococcus aureus.
Therefore, treatment
for these infections consists of a penicillinase resistant
penicillin like dicloxacillin, which covers both group A
beta-hemolytic streptococci and
Staphylococcus aureus.
"Mom, my throat hurts and my skin is disinte-
grating!!!!"
Necrotizing Fasciitis ("Flesh-eating Streptococ-
cus"): This type of group A beta-hemolytic streptococcal
infection has actually been around for years but may in-
deed be on the rise (news coverage certainly is). Certain
strains have M proteins that block phagocytosis, allow-
ing the bacteria to move rapidly through tissue. Strep-
tococci enter through a break in the skin caused by
trauma and then follow a path along the fascia which
lies between the subcutaneous tissue and muscle.
Within a day the patient develops swelling, heat, and
redness that moves rapidly from the initial skin infec-
tion site. A day later the skin color changes from red to
purple to blue, and large blisters (bullae) form. Later
the skin dies and muscle may also become infected
(myositis).
This infection must be recognized early and the fas-
cia surgically removed. Rapid antibiotic therapy is cru-
cial. Group A beta-hemolytic streptococci are still
exquisitely sensitive to penicillin G. It may be wise to
add clindamycin, as this drug rapidly shuts down strep-
tococcal metabolism and will block toxin production
(Holm, 1996; Stevens, 1988). Even with antibiotics and
surgery the mortality rate is high (> 50%).

Figure 4-2
Necrotizing fasciitis can also be caused by Staphylo-
coccus, Clostridium species, gram-negative enterics, or
mixed infection with more than one of these bacteria
( Stevens, 1992).
3) Scarlet fever: Certain beta-hemolytic group A
streptococci not only cause a sore throat, but also pro-
duce an exotoxin called either pyrogenic toxin or ery-
throgenic toxin. This exotoxin is acquired by lysogenic
conversion (see Chapter 3). The exotoxin produces fever
(so it is pyrogenic) and causes a scarlet-red rash. The
rash begins on the trunk and neck, and then spreads to
the extremities, sparing the face. The skin may peel off
in fine scales during healing.
"Mom, my body is turning scarlet!!!!"
Fig. 4-2. "MOM, help!!!" Pharyngitis, impetigo, and
scarlet fever. Note that scarlet fever actually spares
the face.
4) Streptococcal toxic shock syndrome: It is now
clear that beta-hemolytic group A streptococci can cause
toxic shock syndrome like that caused by Staphylococ-
cus aureus. Similar to scarlet fever, streptococcal toxic
shock syndrome is also mediated by the release of pyro-
genic toxin. See Chapter 5 and Fig. 5-9 for more details.
Consider adding clindamycin to penicillin G, as the for-
mer rapidly shuts down streptococcal metabolism and
toxin production (Stevens, 1988; Holm, 1996).
Delayed Antibody-Mediated Disease
1) Rheumatic fever:
With the advent of penicillin, rheumatic fever is now
uncommon. It usually strikes children 5-15 years of age.
When it occurs, it has been shown to follow untreated
beta-hemolytic group A streptococcal pharyngitis (but
CHAPTER 4. STREPTOCOCCI
24
Figure 4-3
NOT after a skin infection). The 6 major manifestations
of rheumatic fever are:
a) Fever.
b) Myocarditis (heart inflammation).
c) Joint swelling (arthritis).
d) Chorea (uncontrolled dance-like movements of the
extremities) which usually begins 2-3 weeks after the
pharyngitis.
e) Subcutaneous nodules (rubbery nodules just un-
der the skin).
f) Rash, called erythema marginatum because it
has a red margin that spreads out from its center.
Fig. 4-3. Picture John Travolta in the movie
Rheumatic Fever, the upcoming sequel to Saturday
Night Fever. His heart is damaged from the stress of
the hours of disco dancing, his joints are aching from
dropping to his knees, and his arms are moving rhyth-
mically in a disco choreiform jam.
Rheumatic fever is antibody-mediated. There are anti-
gens in the heart that are similar to the antigens of the
beta-hemolytic group A streptococci. Therefore, the anti-
bodies that form to eradicate this particular streptococcus
also cross-react with antigens in the heart. This immuno-
logic attack on the heart tissue causes heart inflamma-
tion, called myocarditis. Patients may complain of chest
pain and may develop arrhythmias or heart failure.
Over years, likely after recurrent infections with
streptococci, the heart becomes permanently damaged.
The most frequently damaged site of the heart is the mi-
tral valve, followed by the aortic valve. These damaged
valves may become apparent many years (10-20) after
the initial myocarditis, and can be picked up on physi-
cal exam because they produce heart murmurs. So,
there is an initial myocarditis, and many years
later rheumatic valvular heart disease develops.
These patients are susceptible to recurrent bouts
of rheumatic fever and further heart damage. To pre-
vent further damage to the heart (which is permanent
and irreversible), prophylactic penicillin therapy is re-

quired for much of the patient's life. This will prevent
future beta-hemolytic group A streptococcal infections,
which if they occur will elicit more of the cross-reacting
antibodies.
Once damaged, the heart valves are susceptible to in-
fection by many other types of bacteria. Therefore, pa-
tients with valvular disease need to be given antibiotics
whenever they have a dental or surgical procedure.
Amoxicillin is commonly given.
The joint pain of rheumatic fever is classified as an
acute migratory polyarthritis, which is to say that joint
pains arise at various sites throughout the day and
night. Fortunately, there is no permanent injury to the
joints.
2) Acute post-streptococcal glomerulonephritis:
This is an antibody-mediated inflammatory disease
of the glomeruli of the kidney. It occurs about one week
after infection of either the pharynx OR skin by
nephritogenic ( having the ability to cause glomeru-
lonephritis) strains of beta-hemolytic group A strepto-
cocci. Fortunately, only a few strains of beta-hemolytic
group A streptococci are nephritogenic. Certain anti-
gens from these nephritogenic streptococci induce an
antibody response. The resulting antigen-antibody com-
plexes travel to and are deposited in the glomerular
basement membrane, where they activate the comple-
ment cascade. This leads to local glomerular destruction
in the kidney.
Clinically, a child will show up in your office, and his
mother will complain that his face is puffy. This is
caused by the retention of fluid from his damaged kid-
ney. His urine is darker than normal (tea or coca-cola
colored) due to hematuria (blood in the urine). The child
may also have hypervolemia secondary to fluid reten-
tion, which can cause high blood pressure. Upon further
questioning you may be able to elicit the fact that he had
a sore throat or skin infection a week or so ago. This type
of glomerular disease usually has a good prognosis (es-
pecially in the pediatric population).
"Mom, my urine is tea colored!!!!"
Fig. 4-4.
Acute post-streptococcal glomerulonephritis
causes tea colored urine (hematuria).
GROUP B STREPTOCOCCI
( also called Streptococcus agalactiae)
These streptococci are also beta-hemolytic. When
thinking of group B streptococci, think of group B for
BABY.
About 25% of women carry these bugs vaginally, and
a baby can acquire these bacteria during delivery. These
organisms cause neonatal (< 3 months of age) meningi-
tis, pneumonia, and sepsis.
CHAPTER 4. STREPTOCOCCI
25
Figure 4-4
Neonates with meningitis do not present with a stiff
neck, which is the classic sign seen in adults. Instead,
they display nonspecific signs, such as fever, vomiting,
poor feeding, and irritability. If you even suspect menin-
gitis, you must act rapidly because every minute counts.
Diagnosis of meningitis is made by a lumbar puncture.
Antibiotics are often started prior to the results of the
lumbar puncture if meningitis is suspected. The organ-
isms that must be covered by the antibiotics include Es-
cherichia coli, Listeria monocytogenes, and well as group
B streptococcus. These are the 3 most common patho-
gens associated with meningitis in infants younger than
3 months of age.
Viridans Group Streptococci
( No Lancefield antigen classification. Members
include Streptococcus salivarius, S. sanguis, S. mitis,
S. intermedius, S. mutans, and others.)
This is a big, heterogeneous group of streptococci that
are not identified based on one Lancefield group.
Viridis is the Latin word for green, and most of the
viridans streptococci are alpha-hemolytic, producing
greenish discoloration on blood agar. They are normal
human gastro-intestinal (G.I.) tract flora that are fre-
quently found in the nasopharynx and gingival crevices.
The viridans streptococci cause 3 main types of infec-
tion: dental infections, endocarditis, and abscesses.
1) Dental infections: Some of the viridans strepto-
cocci, especially S. mutans, can bind to teeth and fer-
ment sugar, which produces acid and dental caries
( cavities!!).
2) Endocarditis: Dental manipulations send show-
ers of these organisms into the bloodstream. Subse-
quently, they can implant on the endocardial surface of
the heart, most commonly on a previously damaged
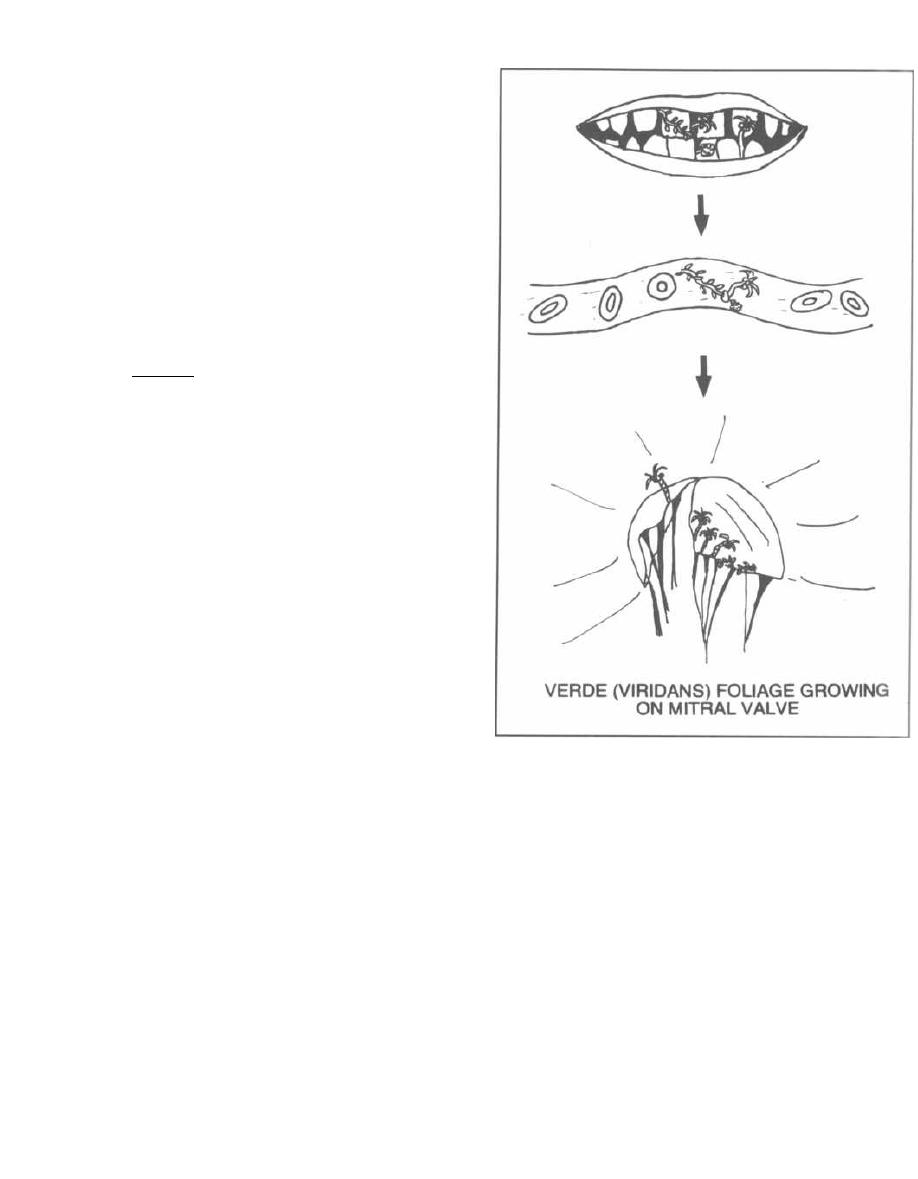
heart valve (such as from old rheumatic fever, a congen-
ital heart defect, or mitral valve prolapse). These bacte-
ria produce an extracellular dextran that allows them to
cling to cardiac valves. This results in subacute bacter-
ial endocarditis (SBE), characterized by a slow (hence
"subacute") growth and piling up of bacteria on the heart
valve (like a pile of bacteria on a petri dish). Clinically,
a patient with subacute bacterial endocarditis slowly de-
velops low-grade fevers, fatigue, anemia, and heart mur-
murs secondary to valve destruction. In contrast, acute
infective endocarditis is caused by a staphylococcal in-
fection, often secondary to IV drug abuse, and is charac-
terized by an abrupt onset of shaking chills, high spiking
fevers, and rapid valve destruction.
Fig. 4-5.
When you think of viridans streptococci,
think ofVERDE, which is the word for "green" in Span-
ish.
Now picture the Verde (green) foliage between
some incisors-you know, palm trees, vines, the works .
When these teeth are pulled by sadistic dentists (they
all are), the Verde foliage enters the blood stream and
settles on leaflets of the heart valves, especially valves
which have been previously damaged (such as valves
damaged by rheumatic fever).
Fig. 4-6. Viridans Streptococcus is eating heart valves
slowly, while
Staphylococcus aureus is
eating fast (No-
tice that these organisms appear as a strip and cluster
respectively!).
Viridans Streptococcus,
slowly eats
away at the valve just as a plant slowly grows into soil.
This is in sharp contrast to
Staphylococcus aureus,
who
received his Olympic gold (aureus) medals for his abil-
ity to rapidly bind to and destroy the heart valves.
Therefore, subacute bacterial endocarditis (SBE) is
caused by viridans Streptococcus, while acute bacterial
endocarditis is the disease associated with
Staphylococ-
cus aureus.
Note that group D streptococci (discussed
below) can also cause subacute bacterial endocarditis.
Interestingly, the streptococci work together as a
team to establish SBE. Initially,
Streptococcus
pyo-
genes
causes rheumatic fever, which damages the heart
valves.
Now, viridans Streptococcus or the group D
streptococci can more easily adhere to the heart valves
and cause SBE!!!
3) Abscesses: There is a subgroup of the viridans
streptococci called the
Streptococcus intermedius
group (comprised of
Streptococcus intermedius, S. con-
stellatus,
and S.
anginosus)
which are microaerophilic
and are part of the normal G.I. tract flora. These oxy-
gen hating critters are often found in abscesses in the
brain or abdominal organs. They are found alone in
pure cultures or in mixed cultures with anaerobes (like
Bacteroides fragilis).
A clinical pearl is that if a
Streptococcus intermedius
group bacteria grows in the blood you should suspect
that there is an abscess hiding in an organ and you
CHAPTER 4. STREPTOCOCCI
26
Figure 4-5
should consider investigating with a CAT scan with
contrast.
Streptococcus InterMediUS:
IMmediately USses (asses) for ABSCESS
GROUP D STREPTOCOCCI
(Enterococci and Non-enterococci)
Traditionally these alpha-hemolytic bacteria have
been divided into two subgroups: the enterococci
(comprised of
Enterococcus faecalis
and
Enterococcus
faecium)
and the non-enterococci (comprised of many
organisms including
Streptococcus bovis
and
Strepto-
coccus equinus).
Recently the enterococci have been
shown to be sufficiently different from the streptococci
to be given their own genus enterococcus.
S.
bovis
and
S. equinus
are still classified as streptococci.

CHAPTER 4. STREPTOCOCCI
Figure 4-6
Enterococcus (faecalis and faecium)
The enterococci take up residence in the human in-
testines and are considered normal bowel flora. They are
alpha hemolytic and unique in that they all grow well in
40% bile or 6.5% NaCl. Clinically, the enterococci are
commonly the infecting agents in urinary tract infec-
tions, biliary tract infections (as they grow well in bile),
bacteremia, and subacute bacterial endocarditis (SBE).
While these bugs are not as virulent as
Streptococcus
pyogenes,
they are always around in the G.I. tract and
prey on weak hospitalized patients. In fact, the entero-
cocci are close to the second most common cause of noso-
comial (hospitally acquired) infections in the U.S. today!
NEWS FLASH!!!! Read all about it! Enterococcus
now resistant to ampicillin and vancomycin!
The enterococci are resistant to most of the drugs we
use to kill gram positive bacteria. We usually treat ente-
rococcal infections with ampicillin plus an aminoglyco-
side. However, many enterococcal strains are now
resistant to both of these agents; in these cases we treat
with vancomycin (see Fig. 16-17). Now our worst night-
mare has been realized: vancomycin resistant enterococci
(VRE) have developed and have been spreading in the
U.S. The resistance property is carried on a gene that is
transferable. Enterococci with this resistance gene alter
their cell wall dipeptide d-alanine-d-alanine (the target
for vancomycin, see page 141), changing it to d-alanine-
lactate. This change prevents vancomycin binding.
The treatment of multiply resistant enterococci is very
difficult and will involve complicated susceptibility test-
ing and infectious disease specialty consultation, and will
require us to take some old and some new antibiotics off
the shelves that are sometimes active against VRE: the
glycopeptides (like vancomycin), teicoplanin, rifampin,
2 7
ciprofloxacin, chloramphenicol, and doxycycline. A newer
class of drugs, the pristinomycins, may also be used:
dalfopristin (Synercid) + quinupristin (RP 59500). These
cause painful arthralgias in 2% and venous irritation
(less with a central line) in 5%. They are currently
available for compassionate use against VRE (Rhone-
Poulenc-Rorer, telephone 610-454-3071). To avoid further
emergence of this resistant strain and worse yet, the
transfer of the genes to more virulent bugs like
Staphylo-
coccus aureus,
we must limit the use of vancomycin. For
example, metronidazole must be used for
Clostridium dif-
Ficile
pseudomembranous colitis instead of vancomycin.
Non-Enterocci
(Streptococcus bovis
and
equinus)
Like the enterococci,
Streptococcus
bovis is hardy,
growing in 40% bile (but not in 6.5% NaCl). It lives in
the G.I. tract, and it causes similar diseases.
An important unique property is that there is a re-
markable association between S.
bovis
infection and
colon cancer!!! In some series 50% of people with S.
bo-
vis bacteremia have a colonic malignancy.
We do not
know if S.
bovis is
a cause of colon cancer or just a
marker of the disease.
BOVIS in the BLOOD:
Better Beware, CANCER in the BOWEL
Streptococcus pneumoniae
(Alias the pneumococcus; No Lancefield antigen)
The pneumococcus is a very important organism be-
cause it is a major cause of bacterial pneumonia and
meningitis in adults, and otitis media in children. pneu-
mococcus is to parents what group B streptococcus is to
Babies.

The pneumococcus does not have Lancefield antigens!
Under the microscope, they appear as lancet-shaped
gram-positive cocci arranged in pairs (diplococci).
The major virulence factor of the pneumococcus is its
polysaccharide capsule, which protects the organism
from phagocytosis. Fortunately, the capsule is anti-
genic, and antibodies specific for the capsule can neu-
tralize the pneumococcus. The only problem is that
there are 84 different capsule serotypes, so surviving an
infection with this organism only provides immunity to
1 out of the 83 possible capsule types.
There are 2 important lab tests to identify the
pneumococcus:
1) Quellung reaction: When pneumococci on a slide
smear are mixed with a small amount of antiserum
(serum with antibodies to the capsular antigens) and
methylene blue, the capsule will appear to swell. This tech-
nique allows for rapid identification of this organism.
2) Optochin sensitivity. Streptococcus pneumoniae
is alpha-hemolytic (partial hemolysis-greenish color)
but Streptococcus viridans is also alpha-hemolytic! To
differentiate the two, a disc impregnated with optochin
(you don't want to know the real name) is placed on the
agar dish. The growth of Streptococcus pneumoniae will
be inhibited, while Streptococcus viridans will continue
to grow.
Streptococcus pneumoniae is the most common cause
of pneumonia in adults. Pneumococcal pneumonia oc-
curs suddenly, with shaking chills (rigors), high fevers,
chest pain with respirations, and shortness of breath.
The alveoli of one or more lung lobes fill up with white
blood cells (pus), bacteria, and exudate. This is seen on
the chest X-ray as a white consolidated lobe. The patient
will cough up yellow-green phlegm that on Gram stain
reveals gram-positive lancet-shaped diplococci.
Fig. 4-7.
The "pneumococcal warrior." He is a mighty
foe, with "capsule" armor, a lung emblem on his shield,
and a lancet-shaped diplococcus lance. The lung em-
blem on his shield shows the severe lobar pneumonia
caused by this organism. Note the consolidation of the
middle right lobe and the lower left lobe, which accom-
pany fever and shaking chills.
Streptococcus pneumoniae is also the most common
cause of otitis media (middle ear infection) in chil-
dren and the most common cause of bacterial meningi-
tis in adults. The classic sign of meningitis, nuchal
rigidity (a stiff neck) is usually present in an adult with
meningitis.
Fig. 4-8.
Otitis media (in children mostly): The pneu-
mococcal warrior's lance zips through the ears of an en-
emy soldier!!
Fig. 4-9.
Meningitis: Our warrior is smashing his en-
emy's head with a hammer!!
CHAPTER 4. STREPTOCOCCI
The first pneumococcal vaccine (the pneumovax) has
25 of the most common capular polysaccharide antigens.
It is given to people for whom pneumococcal pneumonia
would be exceptionally deadly, such as immunocompro-
mised or elderly folk. Individuals without spleens (as-
plenic) or with HIV disease are unable to defend
themselves against encapsulated bacteria and should
also be vaccinated. Unfortunately, the polysaccharide
vaccine has low immunogenicity and efficacy in chil-
dren. A new heptavalent conjugate vaccine contains 7
(thus heptavalent) capsular polysaccharide antigens
from serotypes 4, 6B, 9V, 14, 18C, 19F, and 23F conju-
gated with a non-toxic diphtheria-toxin protein, to in-
crease its immunogenicity. This vaccine has almost
100% efficacy in the prevention of invasive pneumococ-
cal infections in children!!! Because serotypes 3, 6B, 9V,
14, 19F, and 23F are the most common causes of otitis
media (bold serotypes are covered by vaccine), it also has
been shown to reduce cases of otitis media in children
( Eskola, et. al. N Engl J Med 2001; 344:403-9).
Figure 4-7
Figure 4-8
Fig. 4-10. We see the doctor shooting a hole through
our warrior (the pneumococci) with the antibody tipped
pneumovax (pneumococcal pneumonia vaccine).
NEWS FLASH!!!! Read all about it!
Streptococcus
pneumoniae
now resistant to penicillins!
Certain strains of
Streptococcus pneumoniae
are now
showing intermediate level resistance to penicillin (min-
imal inhibitory concentrations (MIC) of 0.1-1.0 micro-
grams penicillin per ml blood) and even high level
resistance (MIC > 2.0 micrograms/ml blood). In some
European countries 2/3 of strains have intermediate or
high level resistance! In the U.S. about 10% of strains
have intermediate resistance and 1% high level resis-
tance; the percentage is much higher in day care set-
tings where children are frequently given antibiotics.
Worse yet, the pneumococcus is also acquiring resis-
tance to erythromycin, trimethoprim/sulfamethoxazole,
and chloramphenicol.
The good news is that high dose penicillin (1 million
units every 4 hours) and the cephalosporins are effec-
tive against bugs with intermediate level resistance. In
areas where high level resistant strains are common
vancomycin will have to be added.
Figure 4-10
CHAPTER 4. STREPTOCOCCI
2 9
Figure 4-9
Unfortunately, we are witnessing dramatic changes in
the way we treat this common and dangerous critter.
Fig. 4-11.
Summary of streptococcal groups.
References
Davies HD, McGreer, A, et al. Invasive group A streptococcal
infections in Ontario, Canada. N. Eng. J. Med. 1996; 335:
547-54.
Eskola J, Kilpi T, et. al. Efficacy of a pneumococcal conjugate vac-
cine against otitis media. N Engl J Med 2001; 344:403-409.
Holm SE, Invasive group A streptococcal infections. N. Eng. J.
Med. 1996; 335: 590-591.
Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice
of Infectious Diseases; 4th edition. New York: Churchill Liv-
ingstone, 1995;1784-1865.
Stevens DL. Invasive group A streptococcus infections. Clinical
Infectious Diseases 1992; 14:2-13.
Stevens DL, Gibbons AE, et al. The eagle effect revisited: effi-
cacy of clindamycin, erythromycin, and penicillin in the treat-
ment of streptococcal myositis. J. Infect. Dis. 1988;158:23-8.
Tuomanen EI, Austrian R, Masure HR. Mechanisms of disease:
pathogenesis of pneumococcal infection. N Engl J Med
1995;332(19 ):1280-1284.
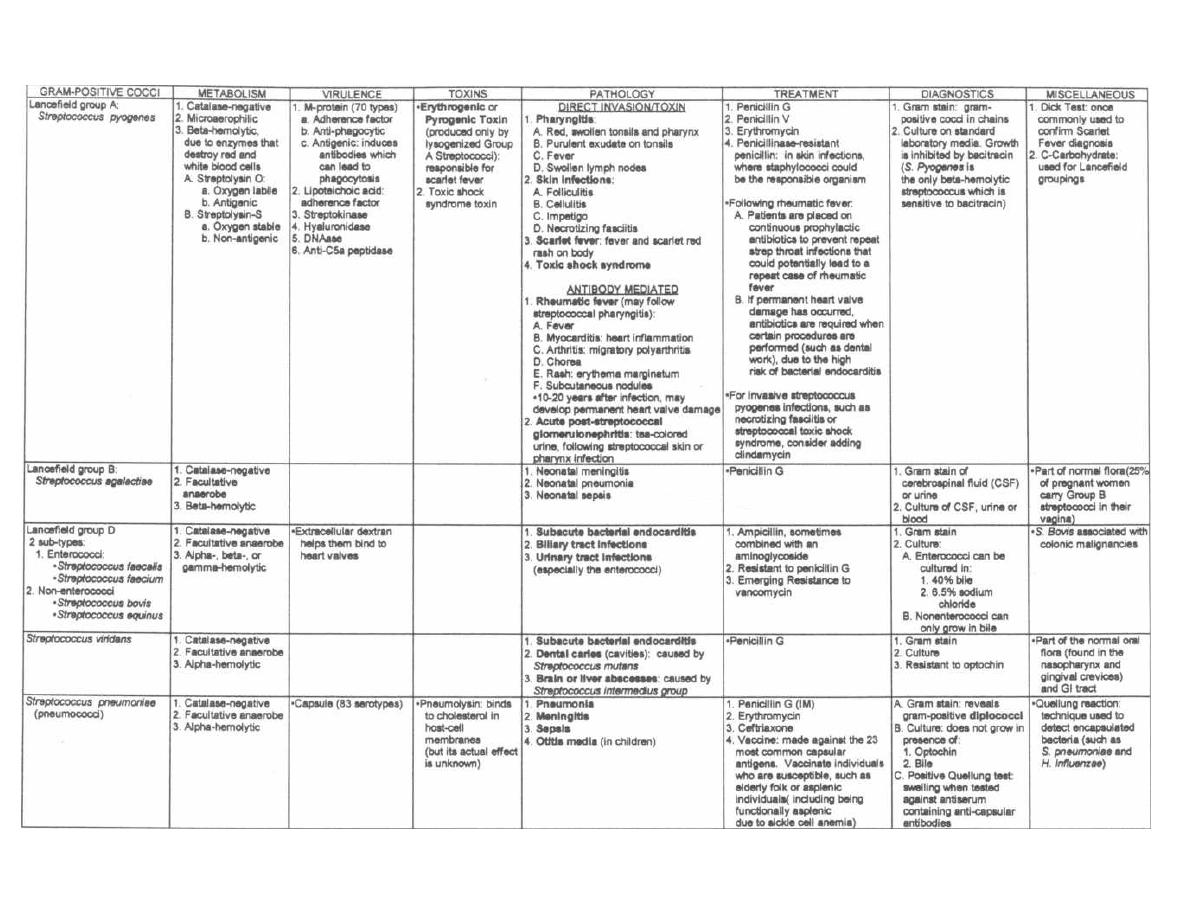
Figure 4-11 STREPTOCOCCI
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple ©MedMaster

Staphylococci are forever underfoot, crawling all over
hospitals and living in the nasopharynx and skin of up
to 50% of people. While at times they cause no symp-
toms, they can become mean and nasty. They will be one
of your future enemies, so know them well.
The 3 major pathogenic species are
Staphylococcus
aureus, Staphylococcus epidermidis,
and
Staphylococ-
cus saprophyticus.
It is extremely important to know how to differ-
entiate staphylococci from streptococci because most
staphylococci are penicillin G resistant! You can do 3
things to differentiate them-Gram stain, catalase test,
and culture.
Fig. 5.1. Staphylococci lie in grape-like clusters as
seen on Gram stain. Visualize this cluster of hospital
staff posing for a group photo.
Staphylococcus aureus
is
catalase-positive, thus explaining the cats in the group
photo.
Staphylococcus aureus
(aureus means "gold")
can be differentiated from the other beta-hemolytic
cocci by their elaboration of a golden pigment when cul-
tured on sheep blood agar. Notice that our hospital
Staff (Staph) all proudly wear gold medals around
their necks.
2) Catalase test: All staphylococci have the enzyme
catalase (streptococci do not!).
Fig. 5-2. Catalase testing, showing a cluster of staph-
ylococci (catalase-positive) blowing oxygen bubbles. To
test, rub a wire loop across a colony of gram-positive
Figure 5-1
CHAPTER 5. STAPHYLOCOCCI
31
cocci and mix on a slide with H2O2. If bubbles appear,
this indicates that H2O2 is being broken down into oxy-
gen bubbles and water; catalase-positive staphylococci
are present.
3) Culture:
Staphylococcus aureus
and certain
streptococci are beta-hemolytic (completely hemolyze
red blood cells on an agar plate), but
Staphylococcus au-
reus
can be differentiated from the other beta-hemolytic
cocci by their elaboration of a golden pigment on sheep
blood agar.
Now that we can differentiate staphylococci from
streptococci, it is important to know which species of
staphylococcus is the actual pathogen. The key point: Of
the 3 pathogenic staphylococcal species, only
Staphylo-
coccus aureus
is coagulase positive!!! It elaborates the
enzyme, coagulase, which activates prothrombin, caus-
ing blood to clot. In Fig. 5-1, note how all the Gold-
Medalists
( Staphylococcus aureus)
hang out together to
show each other their gold medals. You can think of
them as coagulating together. So when a gram-positive
coccus in clusters is isolated in culture, the microbiology
laboratory will do a coagulase test. If they report to you
that the test demonstrates coagulase
positive
grampos-
itive cocci in clusters you know you have
Staphylococcus
aureus.
If they report coagulase
negative
gram-positive
cocci in clusters, think of
Staphylococcus epidermidis
or
staphylococcus saprophyticus.
Figure 5-2
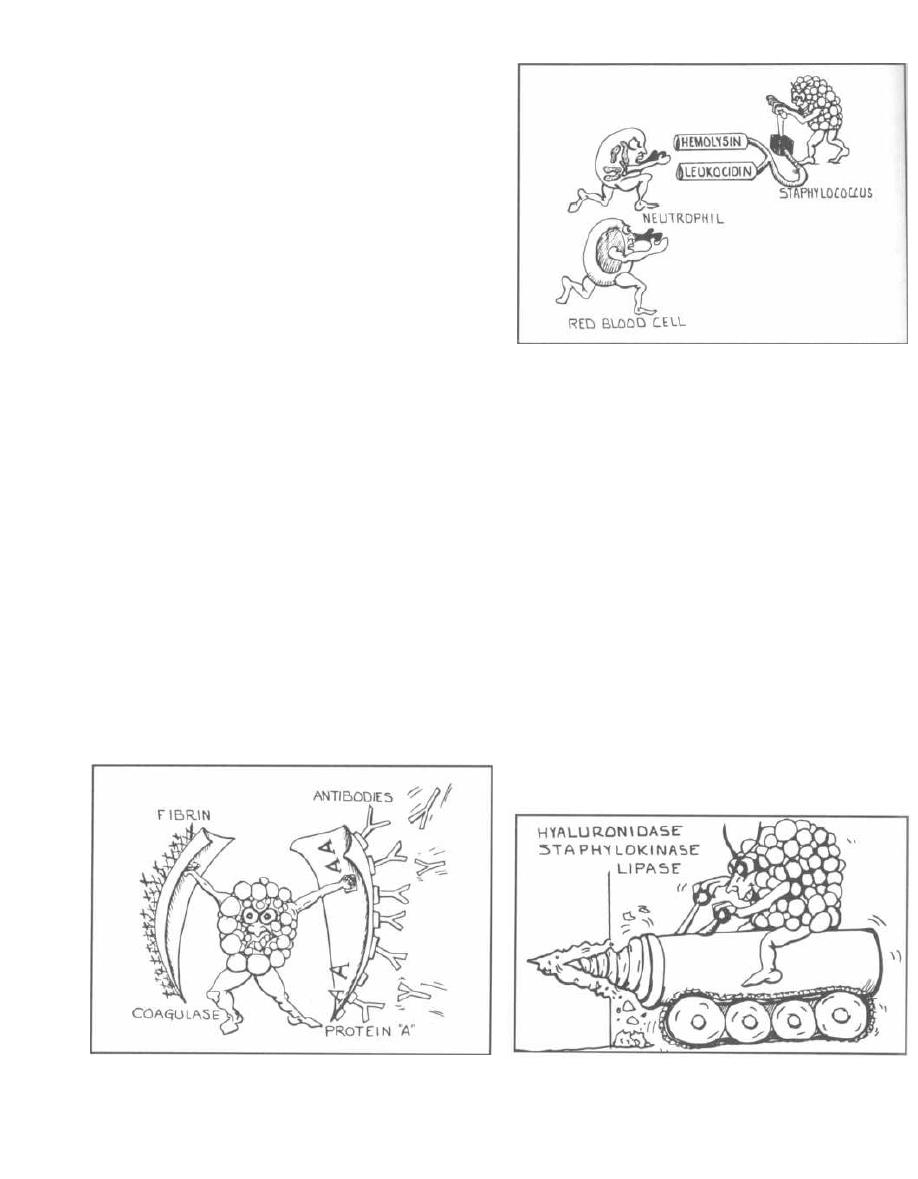
Staphylococcus aureus
This critter has a microcapsule surrounding its huge
peptidoglycan cell wall, which in turn surrounds a cell
membrane containing penicillin binding protein (also
called transpeptidase-see page 114). Numerous pow-
erful defensive and offensive protein weapons stick out
of the microcapsule or can be excreted from the cyto-
plasm to wreak havoc on our bodies:
Proteins That Disable Our
Immune Defenses
1) Protein A: This protein has sites that bind the Fc
portion of IgG. This may protect the organism from op-
sonization and phagocytosis.
2) Coagulase: This enzyme can lead to fibrin forma-
tion around the bacteria, protecting it from phagocytosis.
3) Hemolysins (4 types): Alpha, beta, gamma, and
delta. They destroy red blood cells, neutrophils, macro-
phages, and platelets.
4) Leukocidins: They destroy leukocytes (white
blood cells).
5) Penicillinase: This is a secreted form of beta-
lactamase. It disrupts the beta-lactam portion of the
penicillin molecule, thereby inactivating the antibiotic
(see Chapter 16).
6) Novel penicillin binding protein: This protein,
also called transpeptidase, is necssary for cell wall pep-
tidoglycan formation and is inhibited by penicillin.
Some strains of Staphylococcus aureus have new peni-
cillin binding proteins that are resistant to penicilli-
nase-resistant penicillins and cephalosporins.
Fig. 5-3.
Staphylococcus aureus wielding protein A
and coagulase shields, defending itself from attacking
antibodies and phagocytosis.
Figure 5-3
CHAPTER 5. STAPHYLOCOCCI
32
Figure 5-4
Fig. 5-4.
Hapless red blood cell following a neutrophil,
running to destruction at the hands of Staphylococcus
aureus and its hemolysis and leukocidin dynamite.
Proteins to Tunnel Through Tissue
1) Hyaluronidase ("Spreading Factor"): This pro-
tein breaks down proteoglycans in connective tissue.
2) Staphylokinase: This protein lyses formed fibrin
clots (like streptokinase).
3) Lipase: This enzyme degrades fats and oils,
which often accumulate on the surface of our body. This
degradation facilitates Staphylococcus aureus' coloniza-
tion of sebaceous glands.
4) Protease: destroys tissue proteins.
Fig. 5-5.
Staphylococcus aureus produces proteins
that allow the bacteria to tunnel through tissue.
Exotoxin Assault Weaponry
1) Exfoliatin: A diffusible exotoxin that causes the
skin to slough off (scalded skin syndrome).
Figure 5-5

Figure 5-6
P
2) Enterotoxins (heat stable): Exotoxins which
cause food poisoning, resulting in vomiting and diarrhea.
3) Toxic Shock Syndrome toxin ( TSST-1): This
exotoxin is analogous to the pyrogenic toxin produced by
Lancefield group A beta-hemolytic streptococci, but is
far more deadly. This exotoxin causes toxic shock
syndrome and is found in 20% of
Staphylococcus
aureaus
isolates. These pyrogenic toxins are called
superantigens and bind to the MHC class II molecules
on antigen presenting cells (such as macrophages). The
toxin-MHC II complex causes a massive T cell response
and outpouring of cytokines, resulting in the toxic shock
syndrome described below. (see Fig. 5-9).
Fig. 5-6.
Staphylococcus aureus
produces exotoxin as-
sault weaponry.
Staphylococcus aureus
causes a broad range of hu-
man disease, and can infect almost any organ system.
The diseases can be separated into 2 groups:
Disease caused by exotoxin release:
1) Gastroenteritis (food poisoning).
2) Toxic shock syndrome.
3) Scalded skin syndrome.
Disease resulting from direct organ invasion by the
bacteria:
1) Pneumonia
2) Meningitis
3) Osteomyelitis
4) Acute bacterial endocarditis
5) Septic arthritis
6) Skin infections
7) Bacteremia/sepsis
8) Urinary tract infection
Diseases Caused by Exotoxin Release
1) Gastroenteritis: Staphylococci can grow in food
and produce an exotoxin. The victim will then eat the
food containing the pre-formed toxin, which then stim-
ulates peristalsis of the intestine with ensuing nausea,
CHAPTER 5. STAPHYLOCOCCI
33
vomiting, diarrhea, abdominal pain, and occasionally
fever. The episode lasts 12 to 24 hours.
Fig. 5-7.
Staphylococcus aureus
gastroenteritis. "I
told you not to eat the mayonnaise, sweetheart!"
2) Toxic Shock Syndrome: You may have heard
about toxic shock syndrome and super-absorbent tam-
Figure 5-7
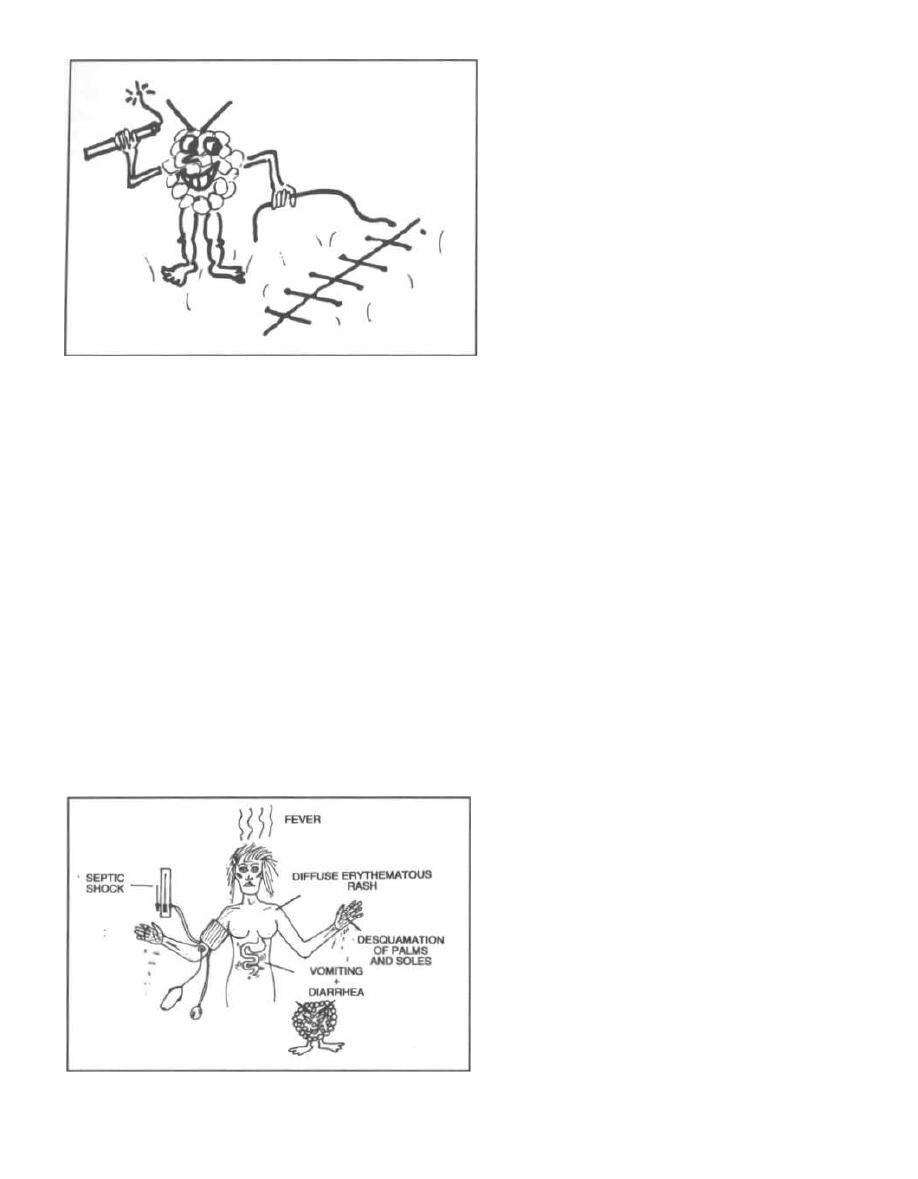
Figure 5-8
pons. It now appears that these tampons, when left in
place for a long time, in some way stimulate Staphylo-
coccus aureus to release the exotoxin TSST-1. This exo-
toxin penetrates the vaginal mucosa and is a potent
stimulator of both tumor necrosis factor (TNF) and
interleukin-1 (see page 15). TSST-1 also dramatically
enhances susceptibility to endotoxin. Tampon use is not
the only cause of this syndrome, since men and non-
menstruating woman can also be affected.
Fig. 5-8. Infected sutures in surgical wounds, cuta-
neous and subcutaneous infections, and infections fol-
lowing childbirth or abortion can all be foci from which
Staphylococcus aureus can release its TSST-1 exotoxin.
Fig. 5-9. Toxic shock syndrome is caused by Staphy-
lococcus aureus releasing TSST-1. This toxin creates
symptoms that you can think of as a hybrid between
food poisoning (enterotoxins) and the streptococcal
pyrogenic toxin that produces scarlet fever. The syn-
drome involves the sudden onset of high fever, nausea,
Figure 5-9
CHAPTER 5. STAPHYLOCOCCI
34
vomiting, and watery diarrhea (enterotoxin-like syn-
drome), followed in a few days by a diffuse erythema-
tous (red) rash (like scarlet fever). The palms and soles
undergo desquamation (fine peeling of the skin) late in
the course of the illness. The toxic shock syndrome
is also associated with septic shock as described in
Chapter 2: blood pressure may bottom out (frank
shock) and the patient may suffer severe organ system
damage (such as acute respiratory distress syndrome
or acute renal failure).
Treatment includes cleaning the infected foci, re-
moval of the tampon or drainage of an infected wound,
along with supportive care. Antibiotics can help by
killing the bacteria and preventing more exotoxin from
being produced. However, antibiotics are not curative
because it is the exotoxin, not the bacteria, which causes
the clinical manifestations.
3) Staphylococcal Scalded Skin Syndrome: This
disease is similar in pathogenesis to toxic shock syn-
drome. A Staphylococcus aureus strain, which produces
exfoliatin toxin, establishes a localized infection and re-
leases a diffusible toxin that exerts distant effects. Un-
like toxic shock syndrome, it usually affects neonates
with local infection of the recently severed umbilicus or
older children with skin infections. Clinically, it causes
cleavage of the middle epidermis, with fine sheets of
skin peeling off to reveal moist red skin beneath. Heal-
ing is rapid and mortality low. The doctor must rule out
a drug allergy, since this can present similarly and may
result in death if the use of the offending drug is not
halted.
Disease Resulting from Direct
Organ Invasion
Fig. 5-10. Diseases caused by direct organ invasion by
Staphylococcus aureus. Visualize the Staph-wielding
wizard. (Note the cluster of staphylococci at the head of
his staff.) The pathology includes:
1) Pneumonia: Staphylococcus aureus is a rare but
severe cause of community-acquired bacterial pneumo-
nia. Pneumonia is more common in hospitalized pa-
tients. It usually follows a viral influenza (flu) upper
respiratory illness, with abrupt onset of fever, chills,
and lobar consolidation of the lung, with rapid destruc-
tion of the lung parenchyma, resulting in cavitations
(holes in the lung). This violent destructive pneumonia
frequently causes effusions and empyema (pus in the
pleural space).
2) Meningitis, Cerebritis, Brain Abscess: These
patients can present with high fever, stiff neck, head-
ache, obtundation, coma, and focal neurologic signs.
3) Osteomyelitis: This is a bone infection that usu-
ally occurs in boys under 12 years of age. The infection
spreads to the bone hematogenously, presenting locally

Figure 5-10
with warm, swollen tissue over the bone and with sys-
temic fever and shakes.
4) Acute Endocarditis: This is a violent destruc-
tive infection of the heart valves with the sudden onset
of high fever (103-105 F°), chills, and myalgias (like a
bad flu). The patient with staphylococcal endocarditis
may have no history of valvular disease and may not
have a murmur. Vegetations grow rapidly on the valve,
causing valvular destruction and embolism of vegeta-
tions to the brain (left heart valve involvement) or lung
(right heart valve infected). Intravenous drug users de-
velop a right-sided tricuspid valve endocarditis and may
present with pneumonia caused by bacterial emboliza-
tion from this infected valve. Endocarditis caused by
Streptococcus viridans
and Group D Streptococci has a
more gradual onset (see Fig. 4-6).
5) Septic Arthritis: Invasion of the synovial mem-
brane
by Staphylococcus aureus
results in a closed in-
CHAPTER 5. STAPHYLOCOCCI
35
fection of the joint cavity. Patients complain of an
acutely painful red swollen joint with decreased range
of motion.
Staphylococcus aureus
is the most common
pathogen causing this disease in the pediatric age
group and in adults over the age of 50. Without prompt
treatment, many patients will permanently lose the
function of the involved joint. Diagnosis requires exam-
ination of the synovial fluid, which will characteristi-
cally appear yellowish and turbid, with a huge number
of neutrophils (>100,000), as well as a positive Gram
stain or culture. Therapy requires drainage of the joint
and antimicrobial therapy.
6) Skin Infections: Minor skin infections are al-
most exclusively caused by either
Streptococcus pyo-
genes
(Group A beta-hemolytic) or
by Staphylococcus
aureus.
It is clinically impossible to differentiate the two
and, in fact, both may be involved. Streptococci can be
treated with penicillin G, but staphylococci are often re-

sistant. Therefore, many doctors believe all skin infec-
tions should be treated with a penicillinase-resistant
penicillin such as dicloxacillin.
Skin infections caused by staphylococci or strepto-
cocci usually follow a major or minor break in the skin,
with scratching of the site spreading the infection:
a) Impetigo: This contagious infection usually oc-
curs on the face, especially around the mouth. Small
vesicles lead to pustules, which crust over to become
honey-colored, wet, and flaky.
b) Cellulitis: This is a deeper infection of the cells.
The tissue becomes hot, red, shiny and swollen.
d) Local Abscesses, Furuncles, and Carbuncles:
An abscess is a collection of pus. Infection of a hair fol-
licle produces a single pus-filled crater with a red rim.
This infection can penetrate deep into the subcutaneous
tissue to become a furuncle. These may bore through
to produce multiple contiguous, painful lesions commu-
nicating under the skin called carbuncles. Significant
abscesses must be surgically drained.
e) Wound infections: Any skin wound can be in-
fected with
Staphylococcus aureus,
resulting in an ab-
scess, cellulitis, or both. When a sutured post-surgical
wound becomes infected, it must be reopened and often
left open to heal by secondary intention (from the bot-
tom of the wound outward).
7)
Blood and catheter infections: Staphylococ-
cus aureus can migrate from the skin and colonize cen-
tral venous catheters resulting in bacteremia, sepsis,
and septic shock (see page 12), as well as endocarditis.
Methicillin-Resistant
Staphylococcus
aureus (MRSA)
Most staphylococci are penicillin-resistant because
they secrete penicillinase. Methicillin, Nafcillin, and
other penicillinase-resistant penicillins are not broken
down by penicillinase, thus enabling them to kill most
strains of
Staphylococcus aureus.
MRSA is a strain of
Staphylococcus aureus
that has acquired multi-drug
resistance, even against methicillin and nafcillin. These
strains tend to develop in the hospital, where broad-
spectrum antibiotics are used. These antibiotics apply
a selection pressure that favors multi-drug resistance in
bacteria (usually acquired by plasmid exchange-see
page 20).
MRSA is transferred from patient to patient by the
hand contact of health care workers, a strong argument
for zealous hand-washing habits!!! This feared bacteria
is resistant to almost all antibiotics. Vancomycin is
one of the few antibiotics useful in treating infections
caused by MRSA, although organisms resistant even to
vancomycin have been reported in the U.S. and Japan
(MMWR
1997; 46: 813-5).
CHAPTER 5. STAPHYLOCOCCI
36
The Centers for Disease Control reports that MRSA in
hospital intensive care units has increased from approx-
imately 20% in
1987
to
60%
in
1997
and MRSA strains
are now increasingly found in the community. Van-
comycin must be used for the treatment of MRSA. Un-
fortunately, strains of
Staphylococcus aureus
resistant
to Vancomycin are now being reported. Hospital use of
vancomycin must be reserved for only critical indica-
tions to protect this antibiotic from emerging resistance.
Staphylococcus epidermidis
This organism is part of our normal bacterial flora
and is widely found on the body. Unlike
Staphylococcus
aureus,
it is coagulase-negative.
This organism normally lives peacefully on our skin
without causing disease. However, compromised hospi-
tal patients with Foley urine catheters or intravenous
lines can become infected when this organism migrates
from the skin along the tubing.
Staphylococcus epidermidis
is a frequent skin conta-
minant of blood cultures. Contamination occurs when
the needle used to draw the blood passes through skin
covered with
Staphylococcus epidermidis.
Drawing blood
from 2 sites will help determine if growth of
Staphylo-
coccus epidermidis
represents a real bacteremic infec-
tion or is merely a contamination. If only one of the
samples grows
Staphylococcus epidermidis,
you can
suspect that this is merely a skin contaminant. How-
ever, if 2 cultures are positive, the likelihood of bac-
teremia with
Staphylococcus epidermidis
is high.
Staphylococcus epidermidis
also causes infections
of prosthetic devices in the body, such as prosthetic
joints, prosthetic heart valves, and peritoneal dialysis
catheters. In fact,
Staphylococcus epidermidis
is the
most frequent organism isolated from infected in-
dwelling prosthetic devices. The organisms have a poly-
saccharide capsule that allows adherance to these
prosthetic materials.
Staphylococcus saprophyticus
This organism is a leading cause (second only to E.
coli)
of urinary tract infections in sexually active young
women. It is most commonly acquired by females
(95%)
in the community (NOT in the hospital). This organism
is coagulase-negative.
Fig. 5-11. Summary chart of staphylococci.
Recommended Review Article:
Lowy FD. Medical progress: Staphylococcus
aureus
infections.
N Engl J Med 1998; 339:520-532.
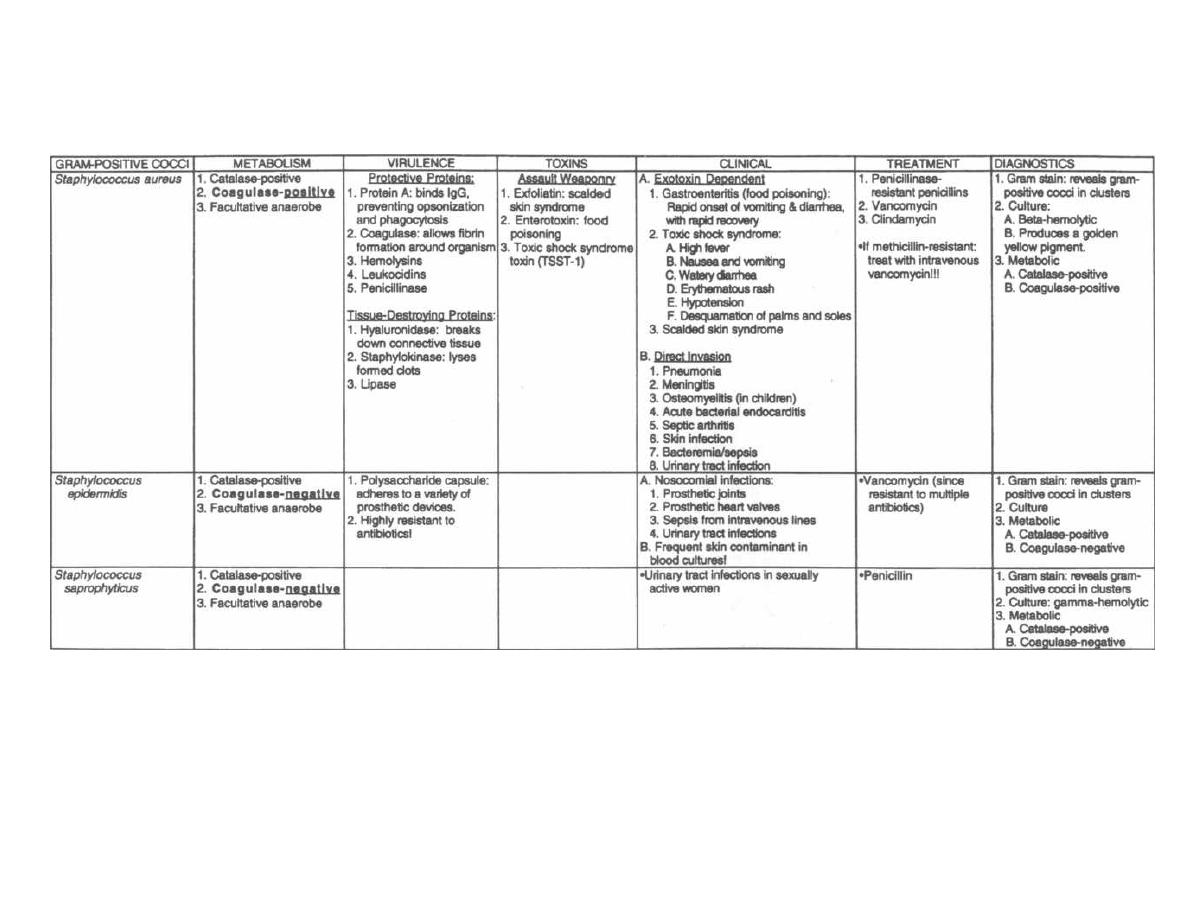
Figure 5-11
STAPHYLOCOCCI
M. Gladwin and B. Trattler,
Clinical
Microbiology Made Ridiculously Simple
©MedMaster
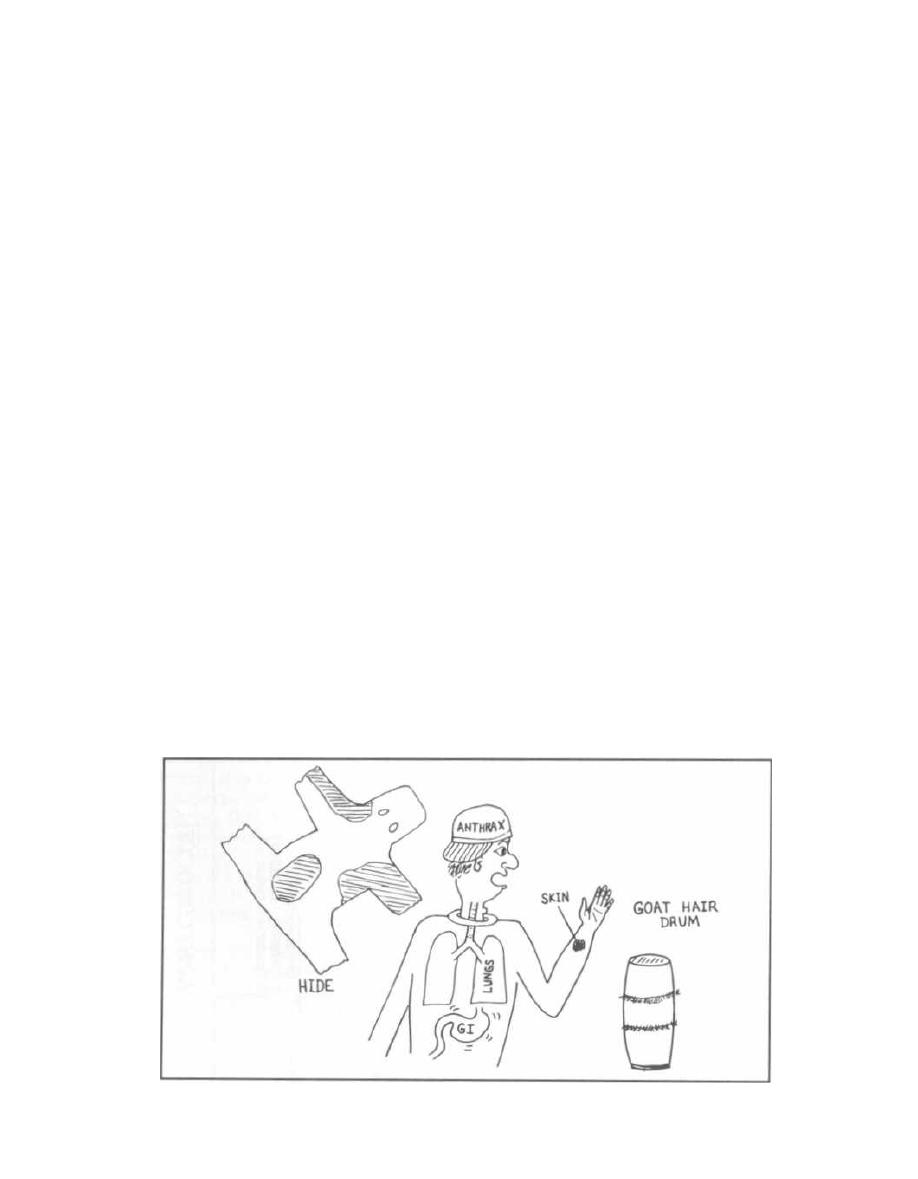
CHAPTER 6.
BACILLUS and CLOSTRIDIUM
(SPORE-FORMING RODS)
There are 6 medically important gram-positive bacte-
ria: 2 are cocci, and 4 are rods (bacilli). Two of the rods
are spore-formers and 2 are not. We have already dis-
cussed the 2 gram-positive cocci (streptococci and
staphylococci). In this chapter we will examine the 2
gram-positive
spore-forming
rods,
Bacillus
and
Clostridium.
Bacillus
and
Clostridium
cause disease by the release
of potent exotoxins (see Fig. 2-8). They differ biochemi-
cally by their like or dislike of oxygen.
Bacillus
enjoys
oxygen (so is aerobic), while Clostridium multiply in
an anaerobic environment. In an air tight Closet, if
you will!
BACILLUS
There are 2 pathogenic species of gram-positive, aer-
obic, spore-forming rods:
Bacillus anthracis
and
Bacil-
lus cereus. Bacillus anthracis
causes the disease
anthrax while
Bacillus cereus
causes gastroenteritis
(food poisoning).
Bacillus anthracis
( Anthrax)
Bacillus anthracis
is unique in that it is the only bac-
terium with a capsule composed of protein (poly-D-
glutamic acid). This capsule prevents phagocytosis.
Bacillus anthracis
causes anthrax, a disease that pri-
marily affects herbivores (cows and sheep). Humans are
exposed to the spores
of
Bacillus anthracis
during direct
contact with infected animals or soil, or when handling
Figure 6-1
38
infected animal products, such as hides or wool. In the
U.S., cases have followed contact with goat hair prod-
ucts from Haiti, such as drums or rugs. Human-to-
human transmission has never been reported.
Bacillus anthracis
forms a spore which is very stable,
resistant to drying, heat, ultraviolet light, and disinfec-
tants, and can survive dormant in the soil for decades.
Once it is introduced into the lungs, intestine, or a skin
wound, it germinates and makes toxins. The germina-
tion and expression of plasmid encoded virulence fac-
tors (on plasmids pXOl and pX02) is regulated by an
increase in temperature to 37°C, carbon dioxide concen-
tration, and serum proteins. So the spore actually acti-
vates only when introduced into the host! Because of its
small size (1-2 µm, ideal for inhalation into alveoli), sta-
bility and the nearly 100% lethality of pulmonary an-
thrax (after spore inhalation), it is considered an ideal
candidate for biological terrorism and warfare. It was
used by the Japanese army in Manchuria in 1940.
Fig. 6-1.
Anthony has Anthrax. This figure demon-
strates how the
Bacillus anthracis
spores are contracted
from contaminated products made of hides and goat hair.
The spores can germinate on skin abrasions (cutaneous
anthrax), be inspired into the lungs (respiratory anthrax),
or ingested into the gastrointestinal tract (GI anthrax).
The spores are often phagocytosed by macrophages in the
skin, intestine, or lung and then germinate, becoming
active (vegetative) gram-positive rods. The bacteria are
released from the macrophage, reproduce in the lym-
phatic system, and then invade the bloodstream (up to
10-100 million bugs per milliliter of blood!!!).

CHAPTER 6. BACILLUS AND CLOSTRIDIUM (SPORE-FORMING RODS)
With a cutaneous anthrax infection (the most com-
mon route of entry),
Bacillus anthracis
rapidly multi-
plies and releases a potent exotoxin. This exotoxin
causes localized tissue necrosis, evidenced by a painless
round black lesion with a rim of edema. This lesion is
called a "malignant pustule" because without antibiotic
therapy (penicillin),
Bacillus anthracis
can continue to
proliferate and disseminate through the bloodstream,
which can cause death. The skin lesion usually resolves
spontaneously in 80-90% of cases, but sometimes
severe skin edema and shock occur.
Pulmonary anthrax, called woolsorter's disease, is not
actually pneumonia. The spores are taken up by
macrophages in the lungs and transported to the hilar and
mediastinal lymph nodes where they germinate. Medi-
astinal hemorrhage occurs resulting in mediastinal
widening (enlarged area around and above the heart seen
on chest radiograph and CT scan) and pleural effusions.
Gastrointestinal anthrax frequently results in death
and fortunately is rare. Outbreaks have followed the in-
gestion of spores (often from contaminated meat).
Bacil-
lus anthracis
matures and replicates within the intestine,
where it releases its exotoxin. The exotoxin causes a
necrotic lesion within the intestine. Patients present with
vomiting, abdominal pain, and bloody diarrhea.
The release of exotoxin is the major reason why an-
thrax carries such a high mortality rate. These toxins
are encoded on a plasmid called pXOl. The exotoxin
contains 3 separate proteins, which by themselves are
nontoxic but together produce the systemic effects of
anthrax:
1) Edema factor (EF) This is the active A subunit
of this exotoxin and is a calmodulin-dependent adeny-
late cyclase. It increases cAMP, which impairs neu-
trophil function and causes massive edema (disrupts
water homeostasis).
2) Protective antigen (PA) promotes entry of EF
into phagocytic cells (similar to a B subunit of the other
A-B toxins, see discussion of exotoxins in Chapter 2).
3) Lethal factor (LF) is a zinc metalloprotease that
inactivates protein kinase. This toxin stimulates the
macrophage to release tumor necrosis factor a and in-
terleukin-1(3, which contribute to death in anthrax.
A second plasmid, pXO2, encodes three genes neces-
sary for the synthesis of a poly-glutamyl capsule. This
capsule inhibits phagocytosis of the vegetative bacteria.
Both plasmids are critical for bacterial virulence.
Rapid identification and prompt use of penicillin, doxy-
cyclin, ciprofloxacin, or levofloxacin are critical in prevent-
ing the high mortality associated with systemic infection by
Bacillus anthracis.
Individuals taking part in high-risk ac-
tivities (petting goats or cows in countries where this dis-
ease is rampant) and military personnel should be given a
39
vaccine composed of the protective antigen (PA). Animals
are vaccinated with living cultures attenuated by the loss
of their antiphagocytic protein capsule. These living vac-
cines are considered too dangerous for human use.
Bacillus cereus
("Be serious")
Bacillus cereus is
different from
Bacillus anthracis
in
that it is motile, non-encapsulated, and resistant to
penicillin.
Bacillus cereus
causes food poisoning (nau-
sea, vomiting, and diarrhea). Food poisoning occurs
when
Bacillus cereus
deposits its spores in food, which
then survive the initial cooking process. The bacteria
then germinate in the food and begin releasing their en-
terotoxin. To inactivate the spores, the cooked food must
be exposed to high temperatures and/or refrigeration.
Bacillus cereus
can secrete 2 types of enterotoxins,
which cause different kinds of food poisoning:
1) A heat-labile toxin similar to the enterotoxin of
cholera and the LT from
Escherichia coli
(see Fig. 2-8)
causes nausea, abdominal pain and diarrhea, lasting
12-24 hours.
2) A heat-stable toxin produces a clinical syndrome
similar to that of
Staphylococcus aureus
food poisoning,
with a short incubation period followed by severe nau-
sea and vomiting, with limited diarrhea.
When a patient is rushed to the hospital with food
poisoning, and examination of the food reveals
B.
cereus,
the best way to respond when your attending or-
ders you to treat the patient with antibiotics is "Be se-
rious, Dr. Goofball." Since the food poisoning is caused
by the pre-formed enterotoxin of
Bacillus cereus,
antibi-
otic therapy will not alter the course of this patient's
symptoms.
CLOSTRIDIUM
Clostridium
are also gram-positive spore-forming
rods. However, they are anaerobic, and can therefore
be separated from the aerobic spore-forming rods
(Bacillus)
by anaerobic culture. This group of bacteria is
responsible for the famous diseases botulism, tetanus,
gas gangrene, and pseudomembranous colitis.
Clostridium
harm their human hosts by secreting ex-
tremely powerful exotoxins and enzymes. Rapid diag-
nosis of a clostridial infection is crucial, or your patient
will die!!!!
Clostridium botulinum
( Botulism)
Clostridium botulinum
produces an extremely lethal
neurotoxin that causes a rapidly fatal food poisoning.
The neurotoxin blocks the release of acetylcholine (ACh)
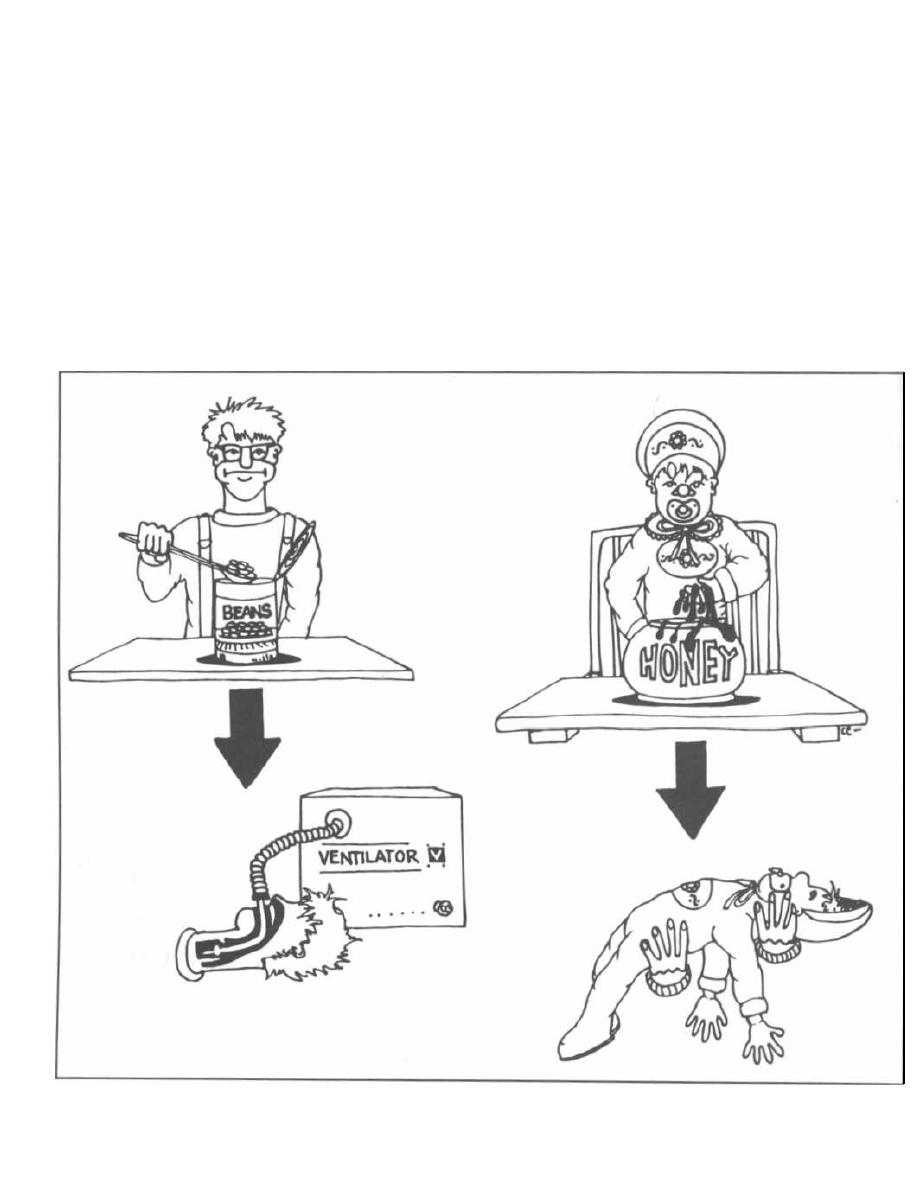
CHAPTER 6. BACILLUS AND CLOSTRIDIUM (SPORE-FORMING RODS)
from presynaptic nerve terminals in the autonomic ner-
vous system and motor endplates, causing flaccid mus-
cle paralysis.
Adult Botulism
Eating smoked fish or home-canned vegetables is as-
sociated with the transmission of botulism.
Clostridium
botulinum
spores float in the air and can land on food.
If the food is cooked thoroughly, the spores will die.
However, if the food with the spores is not cooked suffi-
ciently, and is then placed into an anaerobic environ-
ment (like a glass jar, can, or zip-lock freezer bag),
Clostridium botulinum
matures and synthesizes its
neurotoxin. Those who consume the contents of the jar
when it is opened weeks later will be ingesting the
potent neurotoxin. These afebrile patients initially
develop bilateral cranial nerve palsies causing double
vision (diplopia) and difficulty swallowing (dysphagia).
This is followed by general muscle weakness, which
rapidly leads to sudden respiratory paralysis and death.
Patients must immediately be treated with an anti-
toxin, which can neutralize only the unbound free neu-
rotoxin in the bloodstream. Intubation and ventilatory
support is critical until the respiratory muscles resume
activity.
Figure 6-2
4 0

CHAPTER 6. BACILLUS AND CLOSTRIDIUM (SPORE-FORMING RODS)
Figure 6-3
Infant Botulism
Infant botulism occurs when infants ingest food
contaminated with
Clostridium botulinum
spores
(cases have followed ingestion of fresh honey contami-
nated with spores). The spores germinate and
Clostrid-
ium botulinum
colonizes the infant's intestinal tract.
From this location, botulism toxin is released.
Initially, the infant will be constipated for two to
three days. This is followed by difficulty swallowing and
muscle weakness. These "floppy" babies must be hospi-
talized and given supportive therapy. Prognosis is ex-
cellent, so antitoxin is generally not used.
Fig. 6-2. Botulism. The adult is eating home-canned
beans with neurotoxin while the infant is eating honey
with spores. The adult often requires intubation and
ventilatory support while the baby is merely "floppy."
Clostridium tetani
(Tetanus)
Clostridium tetani
causes tetanus, a disease that
classically follows a puncture wound by a rusty nail but
can follow skin trauma by any object contaminated with
spores.
Clostridium tetani
spores, which are commonly
found in soil and animal feces, are deposited in the
wound and can germinate as long as there is a localized
anaerobic environment (necrotic tissue). From this lo-
cation,
Clostridium tetani
releases its exotoxin, called
tetanospasmin.
The tetanus toxin ultimately causes a sustained con-
traction of skeletal muscles called tetany.
Fig. 6-3. Tetany occurs after the tetanus toxin is
taken up at the neuromuscular junction (end plate) and
is transported to the central nervous system. There the
toxin acts on the inhibitory Renshaw cell interneurons,
preventing the release of GABA and glycine, which are
41
Figure 6-4
inhibitory neurotransmitters. This inhibition of in-
hibitory interneurons allows motor neurons to send a
high frequency of impulses to muscle cells, which re-
sults in a sustained tetanic contraction.
Fig. 6-4. Clinically, the patient with tetanus presents
with severe muscle spasms, especially in the muscles of
the jaw (this is called trismus, or lockjaw). The af-
fected patient exhibits a grotesque grinning expression,
called risus sardonicus, which is due to spasm of the
facial muscles. Mortality is high once the stage of lock-
jaw has been reached.
Because of the high mortality of tetanus, prophylactic
i mmunization with formalin-inactivated toxin (tetanus
toxoid) is performed once every ten years in the U. S.
This booster serves to regenerate the circulating anti-
bodies against tetanus toxin, that were first generated
via childhood immunizations. You may not remember
your first shot (you were probably just 2 months old at
the time), but all children in the U. S. are immunized
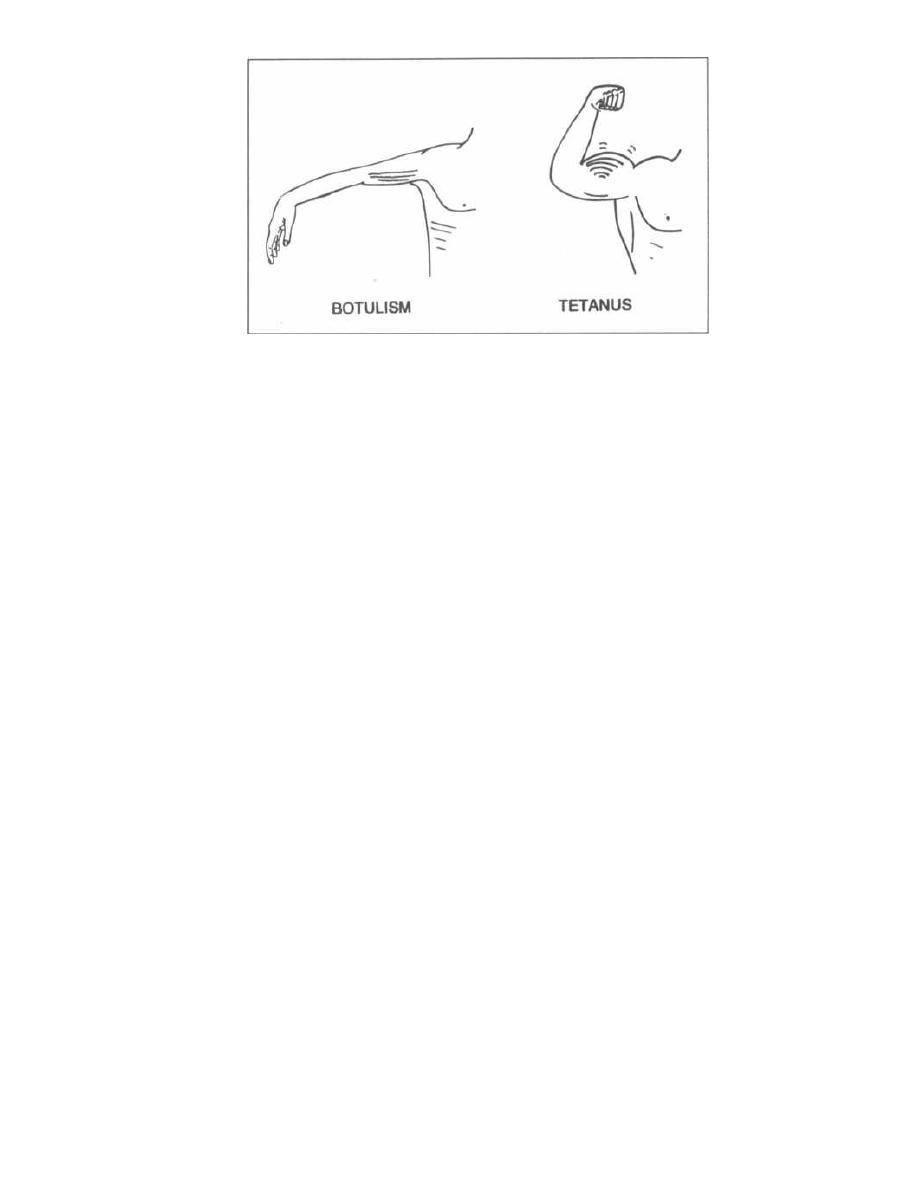
CHAPTER 6. BACILLUS AND CLOSTRIDIUM (SPORE-FORMING RODS)
Figure 6-5
with a series of DPT (diphtheria-pertussis-tetanus)
shots at ages 2, 4, 6, and 18 months, followed by a
booster before entry into school (4-6 years). This regi-
men provides protection from tetanus (along with diph-
theria and pertussis). However, the protection from
tetanus only lasts about 10 years so booster shots of
tetanus are given every 10 years.
In the emergency room you will encounter 3 types of
patients with skin wounds:
1) Patients who were immunized as a child and re-
ceived periodic boosters but the last shot was more
than 10 years ago. These patients are given another
booster.
2) Patients who have never been immunized. Not
only do these patients need a booster, but they should
also receive preformed antibodies to the tetanus toxin
called human tetanus immune globulins.
3) Patients who come to the hospital having already
developed tetanus. The big picture is to clear the toxin
and the toxin-producing bacteria and to keep the pa-
tient alive until the toxin has cleared. This is accom-
plished in the following 5 steps of therapy:
a) Neutralize circulating toxin with human
tetanus immune globulins.
b) Give an immunization booster to stimulate the
patient's own immune system to develop anti-
tetanus toxin antibodies.
c) Clean the wound, excising any devitalized tis-
sue, to remove any remaining source of Clostridium
tetani.
d) Antibiotics (penicillin) may help to clear the
remaining toxin-producing bacteria.
e) Provide intensive supportive therapy until the
toxin is cleared. Muscle relaxants may have to be
administered, and the patient may have to be placed
on a ventilator.
4 2
Fig. 6-5.
Both botulism and tetanus can lead to res-
piratory failure requiring mechanical ventilation. In
botulism, there is flaccid muscle paralysis as the acetyl-
choline at the motor end plate is blocked. In tetanus
there is constant muscle contraction, as the inhibitory
signals are blocked. The tetanic contraction of the res-
piratory muscles also results in respiratory failure.
Clostridium perfringens
( Gas Gangrene)
Everyone has heard of gas gangrene. Prior to anti-
biotics, Clostridium perfringens devastated soldiers
wounded in battle. This bacterium, whose spores can be
found in the soil, matures in anaerobic conditions and
produce gas. The spores can contaminate wounds from
battle or other trauma. Deep wounds with lots of dead
tissue create an anaerobic environment that offers an
excellent home for Clostridium perfringens. As this
anaerobic organism grows, it releases its battery of
exotoxin enzymes (see Fig. 2-8), causing further tissue
destruction.
Clinically, there are 2 classes of infection with Clos-
tridium perfringens:
1) Cellulitis/wound infection: Necrotic skin is
exposed to Clostridium perfringens, which grows and
damages local tissue. Palpation reveals a moist, spongy,
crackling consistency to the skin due to pockets of gas;
this is called crepitus.
2) Clostridial myonecrosis: Clostridium perfrin-
gens, inoculated with trauma into muscle, secretes exo-
toxins that destroy adjacent muscle. These anaerobic
bacteria release other enzymes that ferment carbohy-
drates, resulting in gas formation. A computerized to-
mogram (CT) scan reveals pockets of gas within the
muscles and subcutaneous tissue. As the enzymes de-

CHAPTER 6. BACILLUS AND CLOSTRIDIUM (SPORE-FORMING RODS)
grade the muscles, a thin, blackish fluid exudes from
the skin.
Clostridial myonecrosis is fatal unless identified and
treated very early. Hyperbaric oxygen, antibiotics (such
as penicillin), and removal of necrotic tissue can be life-
saving.
Clostridium difficile
(Pseudomembranous Enterocolitis)
While you may never see a case of anthrax, tetanus,
or botulism in your career, this will certainly NOT
be the case with
Clostridium difficile. You will
tangle
with this critter frequently.
Clostridium difficile is
the
pathogen responsible for antibiotic-associated pseudo-
membranous colitis (diarrhea), which can follow the use
of broad spectrum antibiotics (such as ampicillin, clin-
damycin, and the cephalosporins). These antibiotics can
wipe out part of the normal intestinal flora, allowing the
pathogenic
Clostridium difficile
that is sometimes pre-
sent to superinfect the colon. Once
Clostridium difficile
grows in abundance, it then releases its exotoxins.
Toxin A causes diarrhea, and Toxin B is cytotoxic to the
colonic cells. This disease is characterized by severe di-
arrhea, abdominal cramping, and fever.
Because of
Clostridium difficile
it becomes very
difficile
(difficult) to give patients antibiotics.
Examination by colonoscopy can reveal red inflamed
mucosa and areas of white exudate called pseudo-
membranes on the surface of the large intestine.
Necrosis of the mucosal surface occurs underneath the
pseudomembranes.
When a patient develops diarrhea while on antibi-
otics (or within a month of being on antibiotics),
43
Clostridium difficile
must be considered as a possible
cause. Samples of the stool can be sent to the laboratory
for a
Clostridium difficile
toxin test. Toxin in the stool
confirms the diagnosis.
Treatment includes discontinuing the initial antibi-
otic and administering metronidazole or vancomycin by
mouth. Both antibiotics kill
Clostridium difficile
and
are not absorbed orally into the bloodstream. So
the METRO train and VAN cruise down the gastroin-
testinal (GI) tract, rather than being absorbed, and run
over the hapless
Clostridium difficile
bacteria (see
Chapter 17, Fig. 17-6).
Fig. 6-6. Summary of the Gram-positive spore-
forming rods
References
Ciaran PK, et al. Current Concepts:
Clostridium difficile Col-
itis. NEJM
1994;330:257-262.
Recommended Review Article:
Dixon TC, Meselson M, et al. Medical Progress: Anthrax. N
Engl J Med
1999;341:815-826.
Hogenauer C, et al. Mechanisms and Management of Antibiotic-
Associated Diarrhea. Clinical Infectious Disease
1998;27:
702-710.
Pellizzari R, et al. Tetanus and Botulism Neurotoxins: mecha-
nisms of action and therapeutic uses. Philosophical Trans-
actions of the Royal Society of London
1999;354:259-268.
Petit L. Clostridium perfringens: toxinotype and genotype.
Trends in Microbiology March
1999;7:104.
Shapiro R. Botulism in the United States: A Clinical and Epi-
demiologic Review. Ann Intern Med
1998;129:221-228.
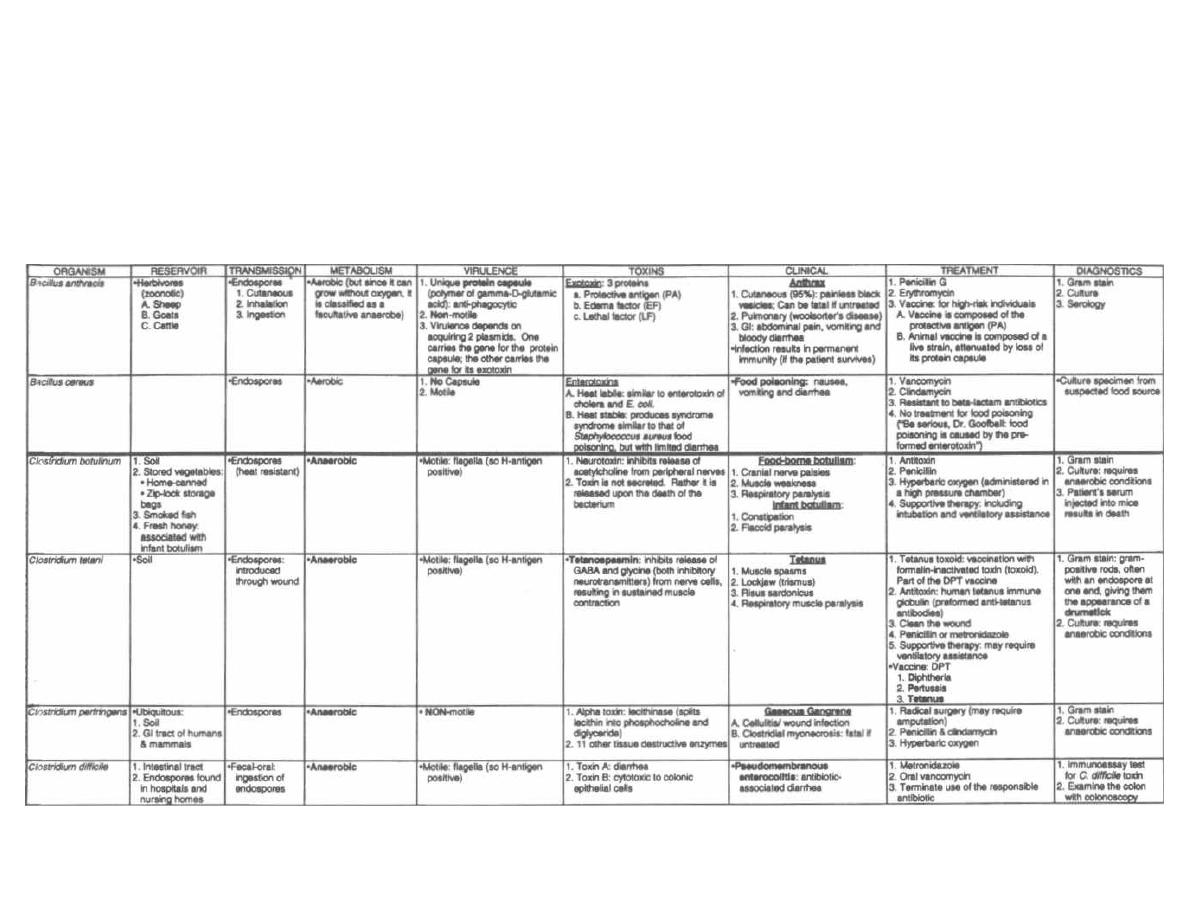
Figure 6-6
GRAM-POSITIVE SPORE-FORMING RODS
M. Gladwin and 8. Trattler, Clinical Microbiology Made Ridiculously Simple aviedMaster

CHAPTER 7.
CORYNEBACTERIUM AND LISTERIA
(NON-SPORE-FORMING RODS)
We have examined the only 2 gram-positive cocci
(Strep-
tococcus
and
Staphylococcus)
and the 2 gram-positive
spore-producing rods
( Bacillus
and
Clostridium). Now
we will discuss the other 2 gram-positive rods (both non-
spore-formers):
Corynebacterium diphtheriae
and
Liste-
ria monocytogenes.
Both of these gram-positive rods
infect patients in the pediatric age group.
CORYNEBACTERIUM DIPHTHERIAE
Corynebacterium diphtheriae is
the pathogen respon-
sible for diphtheria. It colonizes the pharynx, forming a
grayish pseudomembrane composed of fibrin, leuko-
cytes, necrotic epithelial cells, and
Corynebacterium
diphtheriae
cells. From this site, the bacteria release a
powerful exotoxin into the bloodstream, which specifi-
cally damages heart and neural cells by interfering with
protein synthesis.
Fig. 7-1.
Visualize the invading
Corynebacterium
diphtheriae
organisms as a tiny invading army overrun-
ning the throat and building a launching platform on
the pharynx. The army quickly constructs exotoxin rock-
ets. From the safety of their pharynx base, they fire off
deadly rockets to the heart and central nervous system.
While working in the pediatric emergency room, you
see a child with a sore throat and fever. There is a dark
inflammatory exudate on the child's pharynx, which ap-
pears darker and thicker than that of strep throat. Al-
though you may feel the urge to scrape off this tightly
adherent pseudomembrane, you must resist this temp-
tation, because bleeding will occur and the systemic ab-
sorption of the lethal exotoxin will be enhanced.
Being the brightest medical student in the pediatric
emergency room, you immediately recognize that this
child probably has diphtheria. Realizing that you are
dealing with an extremely powerful exotoxin, you
quickly TELL yoUR InTErn not to "loaf around" (Lo-
effler's). Immediately send the throat and nasopharynx
swabs for culture on potassium tellurite agar and
Loeffler's coagulated blood serum media. However,
these culture results will not be ready for days!!! You
may try a Gram stain of a specimen from the pseudo-
membrane, but gram-positive rods are not always seen.
Since there is no time to loaf with diphtheria, it is often
best to proceed rapidly to treatment via the following 3-
step method.
1) Antitoxin: The diphtheria antitoxin only inacti-
vates circulating toxin, which has not yet reached its
45
target tissue, so this must be administered quickly to
prevent damage to the heart and nervous system.
2) Penicillin or erythromycin: Either antibiotic
will kill the bacteria, preventing further exotoxin re-
lease and rendering the patient non-contagious.
3) DPT vaccine: The child must receive the DPT
vaccine, as infection by
Corynebacterium diphtheriae
does not always result in immunity to future infection
by this organism. The DPT vaccine stands for:
D = Diphtheria; P = Pertussis (Whooping Cough); and
T = Tetanus. The diphtheria portion contains formalin
i nactivated diphtheria toxin (see Chapter 6, page 41, for
more on DPT).
Now that therapy has been administered, we can sit
back, relax, and confirm our clinical suspicion of diph-
theria. On the potassium-tellurite plate, colonies of
Corynebacterium diphtheriae
become gray to black
within 24 hours. With Loeffler's coagulated blood
serum, incubation for 12 hours followed by staining
with methylene blue will reveal rod-shaped pleomor-
phic bacteria.
Fortunately for nonimmunized children, not all
Corynebacterium diphtheriae
secrete this exotoxin. Just
as Group A beta-hemolytic streptococci must first be
l ysogenized by a temperate bacteriophage to produce
the erythrogenic toxin that causes scarlet fever,
Corynebacterium diphtheriae
first must be lysogenized
by a temperate bacteriophage which codes for the diph-
theria exotoxin.
This powerful exotoxin contains two subunits. The B
subunit binds to target cells and allows the A subunit to
enter the cell. Once inside the cell, the A subunit blocks
protein synthesis by inactivating elongation factor
( EF2), which is involved in translation of eucaryotic
mRNA into proteins (See Fig. 2-S). Notice an interest-
ing comparison: Anti-ribosomal antibiotics are specifi-
cally designed to inhibit protein synthesis in bacterial
( procaryotic) cells. Similarly, this exotoxin specifically
inhibits protein synthesis in humans (eucaryotes). Thus
this exotoxin can be considered a "human antibiotic,"
because its damage to heart and neural cells can be
lethal.
LISTERIA MONOCYTOGENES
If your attending or professor ever blurts out the silly
statement, "Gram-negative organisms have endotoxin
while gram-positive organisms do not," you can proudly
point out that Listeria monocytogenes, a gram-
positive motile rod, actually has endotoxin! Al-
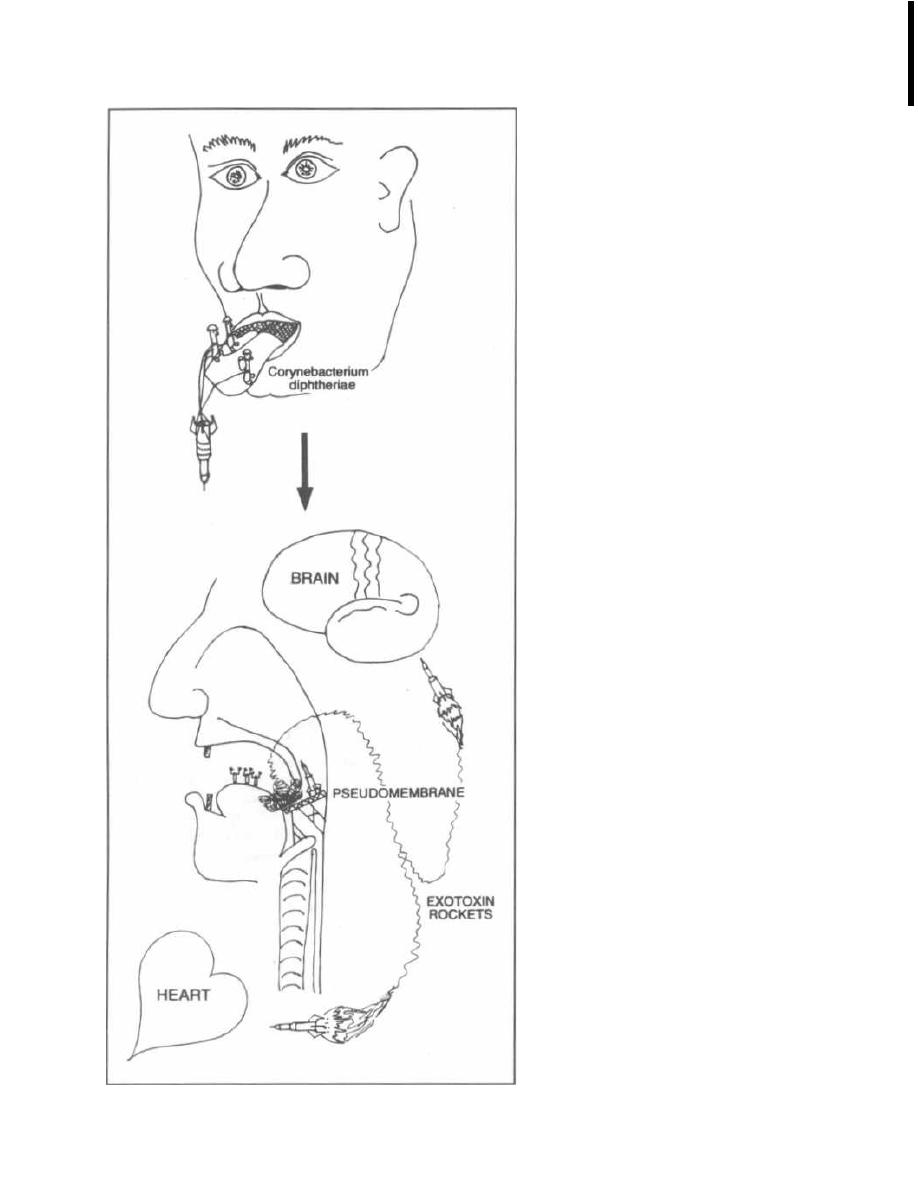
CHAPTER 7. CORYNEBACTERIUM AND LISTERIA (NON-SPORE FORMING RODS)
Figure 7-1
46
though the clinical relevance of this fact is debatable, it
is wonderful for that rare moment when you actually
i mpress your attending.
If your attending, in retaliation, then attempts to
pimp you into submission, describe how Listeria mono.
cytogenes exhibits a tropism for nervous tissue, and thus
is a common cause of meningitis in 2 particular groups.
The first group is neonates, who contract this organism
from their asymptomatic mothers during delivery.
Lis-
teria monocytogenes is the third most common cause of
neonatal meningitis, following only group B streptococci
and Escherichia coli. The second group of patients at
risk for Listeria meningitis is immunosuppressed pa-
tients, such as those with cancer, renal transplants, or
AIDS. The mortality rate for meningitis in this second
group is extremely high.
You may wonder why this organism invades neonates
and certain immunosuppressed patients but not an
immune competent host. The main reason is that Lis-
teria monocytogenes is a resistant fellow, able to hide
out and survive within certain immune cells, such as
macrophages and neutrophils that can phagocytose, or
engulf, foreign objects such as bacteria. Since they can
survive either outside or within cells, Listeria mono-
cytogenes is called a facultative intracellular organ-
ism (see Fig. 2-7). However, in immune competent
hosts, the immune system can release factors that acti-
vate the macrophage, so that these cells can now destroy
the "vagrant" bacteria within them. Immunologists re-
fer to this immune system-mediated method of destroy-
ing Listeria as cell-mediated immunity. However,
neonates (up to 3 months of age) and immuno sup-
pressed patients are unable to activate their phagocytic
cells, thus allowing Listeria monocytogenes to flourish
and infect the meninges. Since pregnancy may also de-
press cell-mediated immunity, Listeria monocytogenes
can infect pregnant women as well, who may develop
meningitis or remain asymptomatic carriers.
Fig. 7-2.
A) The macrophage of a neonate or an im-
munosuppressed patient. B) The macrophage of an im-
mune competent person.
When meningitis develops in a patient who is at high
risk for Listeria monocytogenes, it is important to treat
it empirically with antibiotics that will cover this bac-
terium. After a lumbar puncture confirms that this is a
bacterial meningitis (cerebrospinal fluid analysis reveals
a high number of neutrophils, a high protein level, a low
glucose, and the Gram stain of the cerebrospinal fluid
may demonstrate gram-positive rods), we must add ei-
ther ampicillin or trimethoprim-sulfamethoxazole
to the antibiotic regimen. These are 2 antibiotics that
cover Listeria monocytogenes.
Fig. 7-3.
Summary of the non-spore-forming gram-
positive rods.

CHAPTER 7. CORYNEBACTERIUM AND LISTERIA (NON-SPORE FORMING RODS)
Figure 7-2
47

'gore
7-i NON SPORE-FORMING GRAM-POSITIVE RODS
M Gladwin and B Trattler. Clinical Microbiology Made Ridiculously Simple ©MedMastei

GRAM-NEGATIVE BACTERIA
CHAPTER
8. NEISSERIA
I
NEISSERIA
MENINGITIDIS
1
NEISSERIA SEEN
UNDER THE MICROSCOPE
NEISSERIA
GONORRHOEAE
Figure 8-1
It's time to examine the only pathogenic gram-negative
cocci, Neisseria.
These guys hang out in pairs and are
thus called diplococci. Each coccus is shaped like a kid-
ney bean, and a pair of cocci sticks together with their
concave sides facing each other, almost making the
diplococcus look like a small doughnut.
Two species cause disease in humans: Neisseria
meningitidis and Neisseria gonorrhoeae.
49
I
Fig. 8-1. Meet the 2 pathogenic kidney beans, which
have been removed from the microscope slide. They are
sitting together at the breakfast table. Notice that they
sit facing each other, forming a gram-negative doughnut-
shaped diplococcus. The bean on the left,
Neisseria
meningitidis,
drinks a pot of coffee and becomes very
nervous and irritable (central nervous system ir-
ritation-meningitis). The other pathogenic kidney

bean
is Neisseria gonorrhoeae,
who is a pervert (notice
how he is displaying the latest center-fold pin-up). He
enjoys hanging out on sexual organs and swimming in
"sexual fluids." He causes the sexually transmitted
disease (STD) gonorrhea.
NEISSERIA MENINGITIDIS
Besides causing meningitis,
Neisseria meningi-
tidis
(also called the meningococcus) causes life-
threatening sepsis (meningococcemia).
Virulence factors of the meningococcus include:
1) Capsule: A polysaccharide capsule surrounds the
bacterium and is antiphagocytic, as long as there are no
specific antibodies to coat (opsonize) the bacterium.
Neisseria meningitidis
is classified into serogroups
based on different capsular polysaccharides, which are
antigenic (stimulate a human antibody response).
There are 9 serotypes of meningococcus (designated A,
B, C, D, X, Y, Z, W135, and 29E). Meningitis is caused
by groups A, B, and C.
2) Endotoxin (LPS): The meningococci can release
blebs of endotoxin, which causes blood vessel destruc-
tion (hemorrhage) and sepsis (Chapter 2, page 12). The
blood vessel hemorrhage is seen on the skin as tiny,
round, red dots of hemorrhage called petechiae (a pe-
techial rash). This same hemorrhaging process can
damage the adrenal glands.
3) IgA1 protease: This is only found in pathogenic
species
of Neisseria.
This enzyme cleaves IgA (a type of
antibody) in half.
4) Neisseria meningitidis
can extract iron from hu-
man transferrin via a non-energy requiring mechanism.
Although
Neisseria meningitidis
has all of the above
virulence factors, it usually blends in and becomes part
of the normal flora of the nasopharynx. These individu-
als (about 5% of the population) are called carriers. Car-
riers are lucky, as this asymptomatic nasopharyngeal
infection allows them to develop anti-meningococcal an-
tibodies (this is called natural immunization).
High-risk groups are:
1) Infants aged 6 months to 2 years
2) Army recruits
During pregnancy, maternal protective antibodies
cross the placenta and provide protection to the new-
born, but only for the first few months of life. Infants
will not manufacture their own protective antibodies for
a few years. Therefore, there is a window period (from
age 6 months to 2 years) when infants are extremely
susceptible to a meningococcal infection (meningitis or
septicemia). Note that
Haemophilus influenzae
has a
similar antibody-free window.
A second scenario for an invasive meningococcal in-
fection occurs when army recruits from all over the
CHAPTER 8. NEISSERIA
50
United States are placed together in close quarters and
must survive "boot camp." In this close-knit group, car-
rier rates are greater than
40%.
Each army recruit may
be a carrier of a particular strain of meningococcus that
the other army recruits' immune systems have never
been exposed to, increasing susceptibility to invasive
disease. Further, due to the mentally and physically ex-
hausting training, the immune system's ability to de-
fend itself is weakened.
Meningococcal Disease
Neisseria meningitidis
spreads via respiratory se-
cretions and usually lives asymptomatically in the
nasopharynx. Rarely, the bacteria will invade the blood-
stream (bacteremia) from the nasopharynx, resulting in
meningitis and/or deadly sepsis (called meningococ-
cemia). The classic "clue" to an invasive meningococcal
infection is the appearance of a petechial rash. This
rash is due to the release of endotoxin from the
meningococcus, causing vascular necrosis, an inflam-
matory reaction, and hemorrhage into the surrounding
skin. Note that the diplococci can be seen (Gram stain)
or cultured from biopsies of the petechiae.
1) Meningococcemia: The intravascular multipli-
cation
of Neisseria meningitidis
results in an abrupt on-
set of spiking fevers, chills, arthralgia (joint pains), and
muscle pains, as well as the petechial rash. These pa-
tients usually look acutely ill. Once in the bloodstream,
the meningococci rapidly disseminate throughout the
body. This can lead to meningitis and/or fulminant
meningococcemia.
2) Fulminant meningococcemia (Waterhouse-
Friderichsen syndrome): This is septic shock (see
Chapter 2, page 12). Bilateral hemorrhage into the
adrenal glands occurs, which causes adrenal insuffi-
ciency. Abrupt onset of hypotension and tachycardia oc-
curs, along with rapidly enlarging petechial skin
lesions. Disseminated intravascular coagulation (DIC)
and coma may develop. Death can occur rapidly (6-8
hours).
3) Meningitis: This is the most common form of
meningococcal disease, usually striking infants < 1
year of age. Infants usually display nonspecific findings
of an infection, including fever, vomiting, irritability,
and/or lethargy. A bulging open anterior fontanelle may
be a sign of meningitis in neonates, while slightly older
infants may display a stiff neck, as well as positive
Kernig's and Brudzinski's signs.
The classic petechial skin rash may occur when
meningococcemia occurs in conjunction with meningi-
tis. This allows the physician to make a presumptive
diagnosis of meningococcal meningitis even before per-
forming a diagnostic spinal tap.

Diagnosis involves Gram stain and culture of the
meningococcus from blood, cerebrospinal fluid, or pe-
techial scrapings. Neisseria grow best on blood agar that
has been heated so that the agar turns brown (called
chocolate agar). The classic medium for culturing Neis-
seria is
called the Thayer-Martin VCN media. This is
chocolate agar with antibiotics, which are included to
kill competing bacteria.
V stands for vancomycin, which kills gram-positive
organisms.
C stands for colistin (polymyxin) which kills all
gram-negative organisms (except Neisseria).
N stands for nystatin, which eliminates fungi.
Therefore, only
Neisseria
(both
Neisseria meningi-
tidis and
Neisseria gonorrhoeae)
are able to grow on this
culture medium. The addition of a high concentration of
CO2 further promotes the growth of
Neisseria.
In the laboratory, the differentiation between the
Neisseria
species is based on
Neisseria meningitidis'
ability to produce acid from maltose metabolism, while
Neisseria gonorrhoeae
cannot!
Prompt treatment with penicillin G or ceftriaxone
is required at the first indication of disseminated
meningococcemia. Close contacts of an infected patient
are treated with rifampin. Immunization with purified
capsular polysaccharides from certain strains (groups
A, C, Y, and W135) is currently available and used for
epidemics and in high-risk groups. The group B poly-
saccharide does not induce immunity, so a vaccine is not
available at present.
NEISSERIA GONORRHOEAE
Neisseria gonorrhoeae,
often called the gonococcus,
causes the second most commonly transmitted sexual
disease, gonorrhea (chlamydial infections are slightly
more common).
Virulence factors of the gonococcus include:
1) Pili: Neisseria gonorrhoeae has complex genes
coding for their pili. These genes undergo multiple re-
combinations, resulting in the production of pili with
hypervariable amino acid sequences. These changing
antigens in the pili protect the bacteria from our anti-
bodies, as well as from vaccines aimed at producing an-
tibodies directed against the pili.
The pili adhere to host cells, allowing the gonococ-
cus to cause disease. They also serve to prevent phago-
cytosis, probably by holding the bacteria so close to
host cells that macrophages or neutrophils are unable
to attack.
CHAPTER 8. NEISSERIA
5 1
2) Protein II: This outer membrane protein is also
involved in adherence to host cells.
Gonococcal Disease in Men
A man who has unprotected sex with an infected
person can acquire
a
Neisseria gonorrhoeae
infection.
This organism penetrates the mucous membranes of the
urethra, causing inflammation of the urethra (urethri-
tis). Although some men will remain asymptomatic,
most will complain of painful urination along with a pu-
rulent urethral discharge (pus can be expressed from
the tip of the penis). Both asymptomatic and sympto-
matic men can pass this infection to another sexual
partner.
Possible complications of this infection include epi-
didymitis, prostatitis, and urethral strictures. Fortu-
nately, this disease is easily cured by a small dose of
ceftriaxone.
Gonococcal Disease in Women
Like men, women can also develop a gonococcal ure-
thritis, with painful burning on urination and purulent
discharge from the urethra. However, urethritis in
women is more likely to be asymptomatic with minimal
urethral discharge.
Neisseria gonorrhoeae
also infects the columnar ep-
ithelium of the cervix, which becomes reddened and fri-
able, with a purulent exudate. A large percentage of
women are asymptomatic. If symptoms do develop, the
woman may complain of lower abdominal discomfort,
pain with sexual intercourse (dyspareunia), and a puru-
lent vaginal discharge. Both asymptomatic and sympto-
matic women can transmit this infection.
A gonococcal infection of the cervix can progress to
pelvic inflammatory disease ( PID, or "pus in dere").
PID is an infection of the uterus ( endometritis), fal-
lopian tubes ( salpingitis), and/or ovaries ( oophori-
tis). Clinically, patients can present with fever, lower
abdominal pain, abnormal menstrual bleeding, and
cervical motion tenderness (pain when the cervix is
moved by the doctor's examining finger). Menstrua-
tion allows the bacteria to spread from the cervix to
the upper genital tract. It is therefore not surprising
that over 50% of cases of PID occur within one week
of the onset of menstruation. The presence of an in-
trauterine device (IUD) increases the risk of a cervical
gonococcal infection progressing to PID.
Chlamydia
trachomatis is
the other major cause of PID (see Chap-
ter 12, pages 80-82).
Complications of PID include:
1) Sterility: The risk of sterility appears to increase
with each gonorrhea infection. Sterility is most com-
monly caused by scarring of the fallopian tubes, which

occludes the lumen and prevents sperm from reaching
the ovulated egg.
2) Ectopic pregnancy: The risk of a fetus devel-
oping at a site other than the uterus is significantly
increased with previous fallopian tube inflammation
(salpingitis). The fallopian tubes are the most common
site for an ectopic pregnancy. Again, with scarring down
of the fallopian tubes, there is resistance to normal egg
transit down the tubes.
3) Abscesses may develop in the fallopian tubes,
ovaries, or peritoneum.
4) Peritonitis: Bacteria may spread from ovaries
and fallopian tubes to infect the peritoneal fluid .
5) Peri-hepatitis (Fitz-Hugh-Curtis syndrome):
This is an infection by
Neisseria gonorrhoeae of
the cap-
sule that surrounds the liver. A patient will complain
of
right upper quadrant pain and tenderness. This syn-
drome may also follow chlamydial pelvic inflammatory
disease.
Gonococcal Disease in Both Men
and Women
1) Gonococcal bacteremia: Rarely,
Neisseria gon-
orrhoeae
can invade the bloodstream. Manifestations
include fever, joint pains, and skin lesions (which usu-
ally erupt on the extremities). Pericarditis, endocardi-
tis, and meningitis are rare but serious complications of
a disseminated infection.
2) Septic arthritis: Acute onset of fever occurs
along with pain and swelling of 1 or 2 joints. Without
prompt antibiotic therapy, progressive destruction of
the joint will occur. Examination of synovial fluid usu-
ally reveals increased white blood cells. Gram stain and
culture of the synovial fluid confirms the diagnosis, re-
vealing gram-negative diplococci
within
the white blood
cells. Gonococcal arthritis is the most common kind of
septic arthritis in young, sexually active individuals.
Gonococcal Disease in Infants
Neisseria gonorrhoeae
can be transmitted from a
pregnant woman to her child during delivery, resulting
in ophthalmia neonatorum. This eye infection usu-
ally occurs on the first or second day of life and can dam-
age the cornea, causing blindness. Because neonatal
CHAPTER 8. NEISSERIA
52
Chlamydia
eye infections are also a threa,ythro-
erythromycin eye drops, which are effective against both
Neis-
Nisseria gonorrhoeae
and
Chlamydia,
are given to all
newborns. Gonococcal conjunctivitis can also occur in
adults.
Diagnosis and Treatment
Diagnosis of
Neisseria gonorrhoeae
infection is best
made by Gram stain and culture on Thayer-Martin
VCN medium. Pus can be removed from the urethra by
inserting a thin sterile swab. When this is Gram stained
and examined under the microscope, the tiny doughnut-
shaped diplococci can be seen within the white blood
cells.
In the past, the combination of penicillin G with
probenicid was the regimen of choice. However, there
arose penicillinase-producing gonococcal strains and
now an even tougher strain, with chromosomally-
mediated antibiotic resistance to many antibiotics,
such as tetracycline, erythromycin, and trimethoprim)
sulfamethoxazole. This resistance is mediated by a
block in antibiotic penetration into the bacterial cell.
The current therapy of choice is ceftriaxone, a third
generation cephalosporin (see page 131). Ceftriaxone
will also treat syphilis. If the patient is allergic to
cephalosporins, spectinomycin or ciprofloxacin can be
used as an alternative. The patient should also be treated
at the same time with doxycycline or azithromycin for
Chlamydia trachomatis,
because up to 50% of patients
will be concurrently infected with this beta-lactam-re-
sistant (ceftriaxone included) bacteria.
BRANHAMELLA CATARRHALIS
(formerly called
Neisseria catarrhalis)
This organism is part
of
the normal respiratory flora
but can cause otitis media, sinusitis, bronchitis, and
pneumonia (all respiratory tract illnesses). Pneumonia
usually occurs in patients with lung disease. These bac-
teria produce beta-lactamase and are thus resistant to
penicillin.
Fig. 8-2. Summary
of Neisseria.
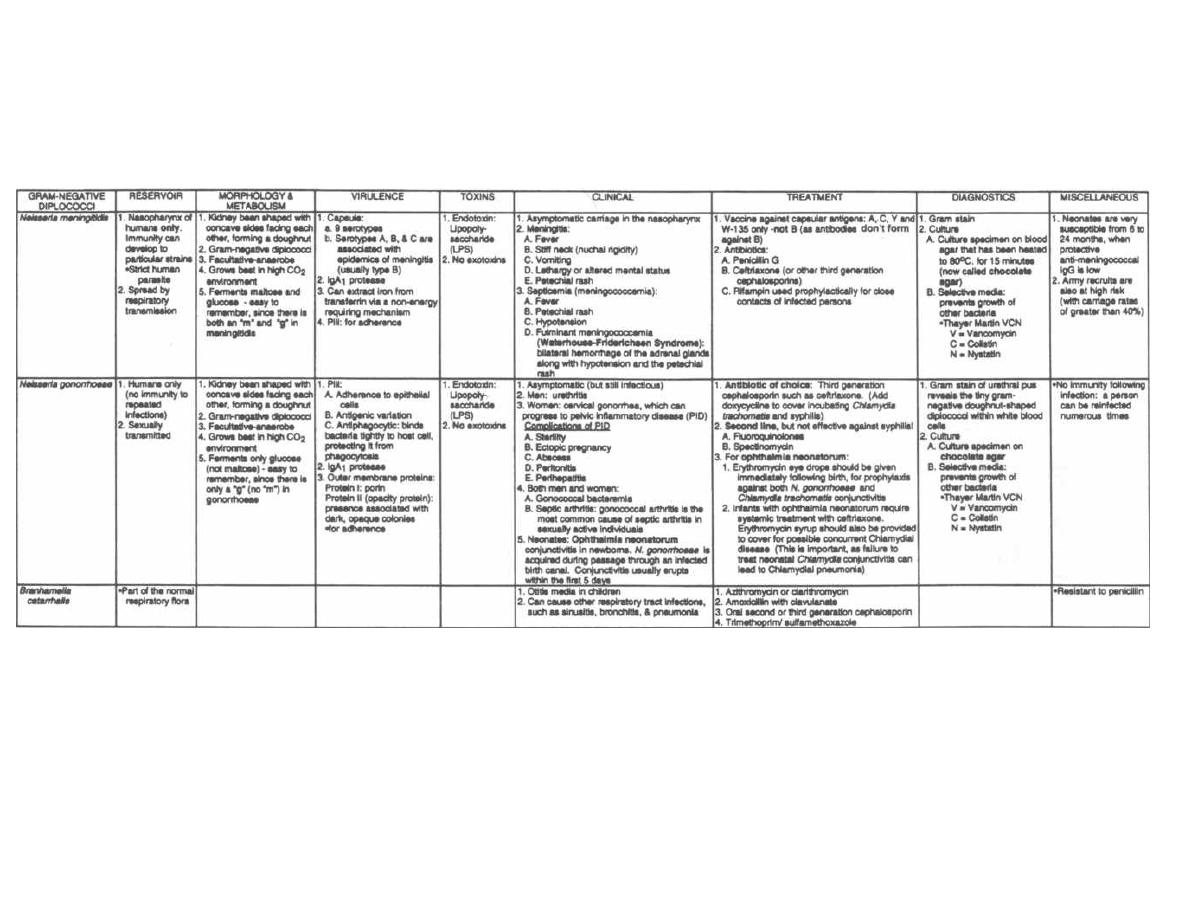
Figure 8-2
NEISSERIA
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
©MedMaster

The enterics are gram-negative bacteria that are part of
the normal intestinal flora or cause gastrointestinal dis-
ease. The family, genus, and species of all the enterics are
organized in the chart at the end of this chapter so that
you will not be confused with the different names. Many
of these bacteria are referred to simply by their genus
name because there are so many different species in some
groups. The main groups are Enterobacteriaceae, Vib-
rionaceae, Pseudomonadaceae and Bateroidaceae.
These organisms are also divided into groups based
upon biochemical and antigenic properties.
Biochemical Classification
Some of the important biochemical properties of the
organisms, which can be measured in the lab, are:
1) The ability to ferment lactose and convert it
into gas and acid (which can be visualized by including
a dye that changes color with changes in pH).
Escher
ichia coli
and most of the enterobacteriaceae ferment
lactose while
Salmonella, Shigella
and
Pseudomonas
aeruginosa
do not.
2) The production of H2S, ability to hydrolyze urea,
liquefy gelatin, and decarboxylate specific amino acids.
Some growth media do 2 things at once: 1) They con-
tain chemicals that inhibit the growth of gram-positive
bacteria that may be contaminating the sample. 2) They
have indicators that change color in the presence of lac-
tose fermentation. The 2 that you should know are:
1) EMB agar (Eosine Methylene Blue): Methylene
blue inhibits gram-positive bacteria, and colonies of lac-
tose fermenters become deep purple to black in this
medium.
Escherichia
coli
colonies take on a metallic
green sheen in this medium.
2) MacConkey agar: Bile salts in the medium in-
hibit gram-positive bacteria, and lactose fermenters de-
velop a pink-purple coloration.
In today's modern laboratories there are plastic trays
with up to 30 different media that measure many bio-
chemical reactions, including those just described. A
colony of unknown bacteria is inoculated onto each
medium and incubated. A computer then interprets the
results and identifies the bacteria.
Fecal Contamination of Water
A classic method for determining whether water has
been contaminated with feces demonstrates some of the
practical uses of biochemical reactions and some impor-
CHAPTER 9. THE ENTERICS
54
tart properties of
Escherichia
coli.
Follow this discus-
sion for the overall big picture.
You are traveling through Uruguay and wind up in a
village whose people are suffering from a terrible diar-
rhea. After giving intravenous fluids to scores of babies,
you start to wonder whether the cause of this infection
can be eradicated. When questioned, the villagers tell
you that they obtain their water from a common river.
You know that the enterics are transmitted by the fecal-
oral route, and you begin to wonder if there is fecal con-
tamination of the river water. How will you prove that
the water is fecally contaminated?
Escherichia coli
to the rescue! You see,
Escherichia
coli
is a coliform, which means that it is a normal in-
habitant of the intestinal tract. Think of
E. coli = coli-
form = colon.
Escherichia
coli
is normally not found
outside the intestine. So if you find
Escherichia coli
in
the village stream water, it does not necessarily mean
that
Escherichia coli
is causing the diarrhea, but it does
tell you that there is fecal matter in the river and that
some enteric may be responsible. You pull out your tat-
tered copy of
Clinical Micro Made Ridiculously Simple
and begin the test.
1) Presumptive Test: You add the river water sam-
ples to test tubes containing nutrient broth (like agar)
that contains lactose. These tubes contain an inverted
vial that can trap gas and a dye indicator that changes
color if acid is produced. You let the sample grow for a
day. If lactose is fermented, gas is produced and the dye
is visualized. You now know that either
Escherichia coli
or a nonenteric bacteria that ferments lactose is in the
water. To find out which you continue...
2) Confirmed Test: Streak EMB agar plates with
the water samples, and the
Escherichia coli
should form
colonies with a metallic green sheen. Also
Escherichia
coli
can grow at 45.5C° but most nonenterics cannot, so
you can grow 2 plates at 45.5C° and 37C° and compare
the colonies on both.
3) Completed Test: Colonies that were metallic
green are placed in the broth again. If they produce acid
and gas, then you know the river water contains
Es-
cherichia coli.
You travel upstream and find an outhouse that has
been built in a tree hanging over the water. You inform
the villagers about the need to defecate in areas that do
not have river runoff and teach them to build latrines.
Within a few weeks the epidemic has ended!
Antigenic Classification
The enterics form many groups, based on cell surface
structures that bind specific antibodies (antigenic de-
Figure 9-1
terminants). The enterics have 3 major surface anti-
gens, which differ slightly from bug to bug.
1) 0 antigen: This is the most external component
of the lipopolysaccharide (LPS) of gram-negative bacte-
ria. The 0 antigen differs from organism to organism,
depending on different sugars and different side-chain
substitutions. Remember O for Outer (see Fig. 1-6, for
more information on LPS).
2) K antigen: This is a capsule (Kapsule) that covers
the 0 antigen.
3) H antigen: This antigenic determinant makes up
the subunits of the bacterial flagella, so only bacteria
that are motile will possess this antigen.
Shigella
does
not have an H antigen.
Salmonella
has H antigens that
change periodically, protecting it from our antibodies.
Fig. 9-1.
The 0 antigen forms the outer part of the cell
membrane, the K antigen wraps around the cell like a cap-
sule, and the arms of the H antigen become wavy flagella.
CHAPTER 9. THE ENTERICS
55
Pathogenesis
The organisms in this chapter produce 2 types of dis-
ease:
1) Diarrhea with or without systemic invasion.
2) Various other infections including urinary
tract infections, pneumonia, bacteremia, and sepsis, es-
pecially in debilitated hospitalized patients.
Diarrhea
A useful concept in understanding diarrhea produced
by these organisms is that there are different clinical
manifestations depending on the "depth" of intestinal
invasion:
1) No cell invasion: The bacteria bind to the in-
testinal epithelial cells but do not enter the cell. Diar-
rhea is caused by the release of exotoxins (called
enterotoxins in the GI tract), which causes electrolyte
and fluid loss from intestinal epithelial cells or epithelial
cell death. Watery diarrhea without systemic symp-
toms (such as fever) is the usual picture. Enterotoxi-
genic
Escherichia coli
and
Vibrio cholera
are examples.
2) Invasion of the intestinal epithelial cells: The
bacteria have virulence factors that allow binding and
invasion into cells. Toxins are then released that de-
stroy the cells. The cell penetration results in a systemic
immune response with local white blood cell infiltration
(leukocytes in the stool) as well as fever. The cell death
results in red blood cell leakage into the stool. Exam-
ples: Enteroinvasive
Escherichia coli, Shigella,
and
Sal-
monella enteritidis.
3) Invasion of the lymph nodes and blood-
stream: Along with abdominal pain and diarrhea con-
taining white and red cells, this deeper invasion results
in systemic symptoms of fever, headache, and white
blood cell count elevation. The deeper invasion can also
result in mesenteric lymph node enlargement, bactere-
mia, and sepsis. Examples:
Salmonella typhi, Yersinia
enterocolitica,
and
Campylobacter jejuni.
Various Other Infections
The enterics are normal intestinal inhabitants and
usually live with us in peaceful harmony. In the hospi-
tal and nursing homes, however, some bad things hap-
pen. They acquire antibiotic resistance and can cause
disease in debilitated patients. They can invade the de-
bilitated patients when Foley catheters are in the ure-
thra or when a patient aspirates vomitus that has been
colonized by the enterics. Because of this hospital ac-
quisition, you will often hear them described as the
hospital-acquired gram-negatives or nosocomial
gram-negatives. Examples:
Escherichia coli, Kleb-

siella pneumoniae, Proteus mirabilis, Enterobacter, Ser-
ratia, and Pseudomonas aeruginosa.
FAMILY ENTEROBACTERIACEAE
Escherichia coli
Escherichia coli normally resides in the colon without
causing disease. However, there is an amazing amount
of DNA being swapped about among the enterics by con-
jugation with plasmid exchange, lysogenic conversion
by temperate bacteriophages, and direct transposon
mediated DNA insertion (see Chapter 3). When Es-
cherichia coli acquires virulence in this manner, it can
cause disease:
Nonpathogenic Escherichia coli (normal
flora) + Virulence factors = DISEASE.
Virulence factors include the following:
1) Mucosal interaction:
a) Mucosal adherence with pili (colonization
factor).
b) Ability to invade intestinal epithelial cells.
2) Exotoxin production:
a) Heat-labile and stable toxin (LT and ST).
b) Shiga-like toxin.
3) Endotoxin: Lipid A portion of lipopolysaccharide
( LPS).
4) Iron-binding siderophore: obtains iron from hu-
man transferrin or lactoferrin.
Diseases caused by Escherichia coli in the presence
of virulence factors include the following:
1) Diarrhea.
2) Urinary tract infection.
3) Neonatal meningitis.
4) Gram-negative sepsis, occurring commonly in de-
bilitated hospitalized patients.
Escherichia
coli Diarrhea
Escherichia coli diarrhea may affect infants or adults.
Infants worldwide are especially susceptible to Es-
cherichia coli diarrhea, since they usually have not yet
developed immunity. Since water lost in the stool is of-
ten not adequately replaced, death from Escherichia
coli diarrhea is usually due to dehydration. About 5 mil-
lion children die yearly from this infection.
Adults (and children) from developed countries, trav-
eling to underdeveloped countries, are also susceptible
to Escherichia coli diarrhea, since they have not devel-
oped immunity during their childhood. This travelers'
diarrhea is the so-called Montezuma's revenge
CHAPTER 9. THE ENTERICS
56
named after the Aztec chief killed at the hands of the
Spanish explorer, Cortez.
The severity of Escherichia coli diarrhea depends
on which virulence factors the strain of Escherichia
coli possesses. We will discuss 3 groups of diarrhea-
producing Escherichia coli. These have been named
based on their virulence factors and the different diar-
rheal diseases they cause.
1) Enterotoxigenic Escherichia coli (ETEC):
This Escherichia coli causes traveler's diarrhea. It has
pili (colonization factor) that help it bind to intestinal
epithelial cells, where it releases exotoxins that are sim-
ilar to the cholera exotoxins discussed on page 62. The
toxins are the heat labile toxin (LT), which is just like
the cholera toxin, and the heat stabile toxin (ST).
These exotoxins inhibit the reabsorption of Na and CL
and stimulate the secretion of Cl
-
and HCO3
-
into the
intestinal lumen. Water follows the osmotic pull of these
ions, resulting in water and electrolyte loss. This pro-
duces a severe watery diarrhea with up to 20 liters be-
ing lost a day!!! The stool looks like rice water just
like cholera!
2) Enterohemorrhagic Escherichia coli (EHEC):
These Escherichia coli also have a pili colonization
factor like the ETEC but differ in that they secrete
the powerful Shiga-like toxin ( also called verotoxin)
that has the same mechanism of action as the Shigella
toxin (see page 58). They both inhibit protein synthesis
by inhibiting the 60S ribosome, which results in
intestinal epithelial cell death. So these bacteria hold
onto the intestinal epithelial cells and shoot away with
the Shiga-like toxin (see Fig. 9-3). The diarrhea is
bloody (hemorrhagic), accompanied by severe abdomi-
nal cramps, and is called hemorrhagic colitis.
Hemolytic uremic syndrome (HUS) with anemia,
thrombocytopenia (decrease in platelets), and renal fail-
ure (thus uremia), is associated with infection by a
strain of EHEC, called Escherichia coli 0157:H7. Nu-
merous outbreaks have occurred secondary to infected
hamburger meat served at fast food chains, suggesting
that cattle may be a reservoir for EHEC.
3) Enteroinvasive Escherichia coli (EIEC): This
disease is the same as that caused by Shigella (page 58).
In fact, the main virulence factor is encoded in a plas-
mid shared by Shigella and Escherichia coli. This plas-
mid gives the bacteria the ability to actually invade the
epithelial cells. EIEC also produces small amounts of
Shiga-like toxin. The host tries to get rid of the invading
bacteria, and this results in an immune-mediated in-
flammatory reaction with fever. White blood cells in-
vade the intestinal wall, and the diarrhea is bloody with
white blood cells. Like shigellosis!
Fig. 9-2. Vibrio cholera, Escherichia coli, and Shigella
dysenteriae all holding hands. Escherichia coli can

Figure 9-2
cause diarrhea indistinguishable from shigellosis and
cholera. The big picture here is that the different types
of diarrhea produced by Escherichia coli and the other
enterics are dependent on virulence acquisition from
plasmids, and there is active sharing of these factors. So
Escherichia coli diarrhea can look just like cholera (rice-
water stools) or just like shigellosis (diarrhea with blood
and white cells).
Escherichia coli Urinary Tract
Infections (UTIs)
The acquisition of a pili virulence factor allows Es-
cherichia coli to travel up the urethra and infect the
bladder (cystitis) and sometimes move further up to in-
fect the kidney itself (pyelonephritis). Escherichia coli
is the most common cause of urinary tract infections,
which usually occur in women and hospitalized patients
with catheters in the urethra. Symptoms include burn-
ing on urination ( dysuria), having to pee frequently
(frequency), and a feeling of fullness over the bladder.
Culture of greater than 100,000 colonies of bacteria
from the urine establishes the diagnosis of a urinary
tract infection.
Escherichia coli Meningitis
Escherichia
coli
is the second most common cause of
neonatal meningitis (group B streptococcus is first).
During the first month of life, the neonate is especially
susceptible.
Escherichia coli Sepsis
Escherichia coli is also the most common cause of
gram-negative sepsis. This usually occurs in debilitated
CHAPTER 9. THE ENTERICS
57
hospitalized patients. Septic shock (see Chapter 2, page
12) due to the lipid A component of the LPS is usually
the cause of death.
Escherichia coli Pneumonia
Escherichia coli is a common cause of hospital-ac-
quired pneumonia.
Klebsiella pneumoniae
This enteric is encapsulated (0 antigen) but is non-
motile (no H antigen). Klebsiella pneumoniae prowls
hospitals, causing sepsis (second most common after
Escherichia coli). It also causes urinary tract infections
in hospitalized patients with Foley catheters. Hospital-
ized patients and alcoholics (debilitated patients) are
prone to
a
Klebsiella pneumoniae pneumonia, which is
characterized by a bloody sputum in about 50% of
cases. This pneumonia is violent and frequently de-
stroys lung tissue, producing cavities. Thick sputum
coughed up with Klebsiella pneumoniae classically
looks like red currant jelly, which is the color of the 0
antigen capsule. The mortality rate is high despite an-
tibiotic therapy.
Proteus mirabilis
This organism is very motile. In fact, when you smear
the bacteria on a plate it will grow not as distinct round
colonies, but rather as a confluence of colonies as the
bacteria rapidly move and cover the plate. This organ-
ism is able to break down urea and is thus often referred
to as the urea-splitting Proteus.
There are 3 strains of Proteus that have cross-reacting
antigens with some Rickettsia (Chapter 12, Fig. 12-11).
They are OX-19, OX-2, and OX-K. This is purely coinci-
dental but serves as a useful clinical tool to determine
if a person has been infected with Rickettsia. Serum
is mixed with these Proteus strains to determine
whether there are antibodies in the serum that react
with the Proteus antigens. If these antibodies are pre-
sent, this suggests that the patient has been infected
with Rickettsia.
Proteus is another common cause of urinary tract in-
fections and hospital-acquired (nosocomial) infections.
Examination of the urine will reveal an alkaline pH,
which is due to Proteus' ability to split urea into NH3
and CO2.
Enterobacter
This highly motile gram-negative rod is part of the
normal flora of the intestinal tract. It is occasionally re-
sponsible for hospital-acquired infections.

Serratia
Serratia is notable for its production of a bright red
pigment. It can cause urinary tract infections, wound
infections, or pneumonia.
Shigella
There are four species of Shigella (dysenteriae,
flexneri, boydii, and sonnei) and all are nonmotile. If
you look back at the picture of Escherichia coli and
Shigella holding hands (Fig. 9-2), you will see that
Shigella has no flagella. Shigella does not ferment lac-
tose and does not produce H2S. These properties can be
used to distinguish Shigella from Escherichia coli (lac-
tose fermenter) and Salmonella (non-lactose fermenter,
produces H2S).
Humans are the only hosts for Shigella, and the
dysentery that it causes usually strikes preschool age
children and populations in nursing homes. Trans-
mission by the fecal-to-oral route occurs via fecally
contaminated water and hand-to-hand contact (Em-
ployees please wash hands!). Shigella is never con-
sidered part of the normal intestinal flora! It is always
a pathogen.
Shigella is similar to enteroinvasive Escherichia coli
( EIEC) in that they both invade intestinal epithelial
cells and release Shiga toxin, which causes cell de-
struction. White cells arrive in an inflammatory reac-
tion. The colon, when viewed via colonoscopy, has
shallow ulcers where cells have sloughed off. The illness
begins with fever (unlike ETEC and cholera, which do
not invade epithelial cells and therefore do not induce
a fever), abdominal pain, and diarrhea. The diarrhea
may contains flecks of bright-red blood and pus (white
cells). Patients develop diarrhea because the inflamed
colon, damaged by the Shiga toxin, is unable to reabsorb
fluids and electrolytes.
Fig. 9-3. Visualize Shazam Shigella with his Shiga
blaster laser, entering the intestinal epithelial cells and
blasting away at the 60S ribosome, causing epithelial
cell death.
Shiga Toxin
This is the same toxin as in EHEC and EIEC, and its
mechanism is the same. There is an A subunit bound to
5 B subunits. The B subunits (B for Binding) bind to the
microvillus membrane in the colon, allowing the entry
of the deadly A subunit (A for Action). The A subunits
inactivate the 60S ribosome, inhibiting protein synthe-
sis and killing the intestinal epithelial cell.
CHAPTER 9. THE ENTERICS
58
Figure 9-3
Salmonella
("The Salmon")
Salmonella is a non-lactose fermenter, is motile (like
a salmon), and produces H2S.
You will hear of Salmonella's Vi antigen. This is a
polysaccharide capsule that surrounds the O anti-
gen, thus protecting the bacteria from antibody attack
on the O antigen. This is just like the K antigen (just to
confuse you!), but with Salmonella they named it Vi
(for virulence).
There are thousands of Salmonella serotypes, but
clinically they are usually divided into three groups:
Salmonella typhi, Salmonella cholerae-suis, and Sal-
monella enteritidis. This will not be that difficult to re-
member because they are named according to the
diseases they cause.
Salmonella differs from the other enterics because it
lives in the gastrointestinal tracts of animals and in-
fects humans when there is contamination of food or wa-
ter with animal feces.
Fig. 9-4. Many animals can carry Salmonella. (Pic-
ture a salmon.) In the U.S. there was even an epidemic
of salmonellosis from pet turtles. Today in the U.S., Sal-
monella is most commonly acquired from eating chick-
ens and uncooked eggs. Salmonella typhi is an
exception as it is not zoonotic (an infectious disease of
Figure 9-4
animals that can be transmitted to man).
Salmonella ty-
phi
is carried only by humans.
Salmonella
(like
Shigella) is
never considered part of
the normal intestinal flora! It is always pathogenic and
can cause 4 disease states in humans: 1) the famous ty-
phoid fever, 2) a carrier state, 3) sepsis, and 4) gas-
troenteritis (diarrhea).
Typhoid Fever
This illness caused by
Salmonella typhi
is also called
enteric fever.
Salmonella typhi
moves one step beyond
EIEC and
Shigella.
After invading the intestinal ep-
ithelial cells, it invades the regional lymph nodes, fi-
nally seeding multiple organ systems. During this
invasion the bacteria are phagocytosed by monocytes
and can survive intracellularly.
So Salmonella typhi is
a facultative intracellular parasite (see Fig. 2-7).
Fig. 9-5. Typhoid fever, caused by
Salmonella typhi,
depicted by a Salmon with fever (thermometer) and rose
spots on its belly. Salmonellosis starts 1-3 weeks after
CHAPTER 9. THE ENTERICS
59
exposure and includes fever, headache, and abdominal
pain that is either diffuse or localized to the right lower
quadrant (over the terminal ilium), often mimicking ap-
pendicitis. As inflammation of the involved organs oc-
curs, the spleen may enlarge and the patient may
develop diarrhea and rose spots on the abdomen-a
transient rash consisting of small pink marks seen only
on light-skinned people.
Diagnose this infection by culturing the blood, urine,
or stool. Ciprofloxacin or ceftriaxone are considered
appropriate therapy.
Figure 9-5
Carrier State
Fig. 9-6. Some people recovering from typhoid fever
become chronic carriers, harboring
Salmonella typhi
in
their gallbladders and excreting the bacteria con-
stantly. These people are not actively infected and do
not have any symptoms. A famous example occurred in
1868 when Typhoid Mary, a Swiss immigrant who
worked as a cook, spread the disease to dozens in New
York City. (Again-employees please wash hands after
using the toilet!) Some carriers actually require surgical
removal of their gallbladders to cure them.
Sepsis
Fig. 9-7. Salmon cruising in the bloodstream to infect
lungs, brain, or bone. This systemic dissemination is
usually caused by
Salmonella choleraesuis
and does not
involve the GI tract.
A pearl of wisdom:
Remember that
Salmonella
is encapsulated with the Vi
capsule. Our immune system clears encapsulated bacte-
ria by opsonizing them with antibodies (see Fig
2-5), and then the macrophages and neutrophils in the
spleen (the reticulo-endothelial system) phagocytose the
opsonized bacteria. So, patients who have lost their
spleens (asplenic), either from trauma or from sickle-cell
disease, have difficulty clearing encapsulated bacteria
and are more susceptible to
Salmonella
infections. Pa-
tients with sickle-cell anemia are particularly
prone to Salmonella osteomyelitis (bone infection).
Vigorous and prolonged antibiotic therapy is required
to treat
Salmonella
osteomyelitis.
Figure 9-7
CHAPTER 9. THE ENTERICS
60
Figure 9-6
Diarrhea (Gastroenteritis)
Fig. 9-8.
Salmonella
diarrhea is the most common
type of
Salmonella
infection and can be caused by any
of hundreds of serotypes of
Salmonella enteritidis.
The
presentation includes nausea, abdominal pain, and di-
arrhea that is either watery or, less commonly, contains
mucous and trace blood. Fever occurs in about half the
Figure 9-S
patients. This diarrhea is caused by a yet-uncharacter-
ized cholera-like toxin (watery diarrhea) and sometimes
also by ileal inflammation (mucous diarrhea).
Treatment usually involves only fluid and electrolyte
replacement, as antibiotics do not shorten the course of
the disease and do cause prolonged bacterial shedding
in the stool. The diarrhea only lasts a week or less.
Yersinia enterocolitica
This motile gram-negative rod is another cause of
acute gastroenteritis. Since entero is part of
Yersinia
enterocolitica's
name, it is not surprising that this or-
ganism is a cause of acute gastroenteritis. It is not re-
CHAPTER 9. THE ENTERICS
61
ally an enteric bacterium but is included here because
it causes diarrhea. This organism is closely related to
Yersinia pestis,
which is the cause of the bubonic plague.
Like
Yersinia pestis,
animals are a major source of
Yersinia enterocolitica; Yersinia enterocolitica
differs in
that it is transferred by the fecal-oral route rather than
the bite of a flea.
Following ingestion of contaminated foods, such as
milk from domestic farm animals or fecally contami-
nated water, patients will develop fever, diarrhea, and
abdominal pain. This pain is often most severe in the
right lower quadrant of the abdomen, and therefore pa-
tients may appear to have appendicitis. Examination of
the terminal ileum (located in the right lower quadrant)
will reveal mucosal ulceration.
The pathogenesis of this organism is twofold:
1) Invasion: Like
Salmonella typhi,
this organism
possesses virulence factors that allow binding to the in-
testinal wall and systemic invasion into regional lymph
nodes and the bloodstream. Mesenteric lymph nodes
swell, and sepsis can develop.
2) Enterotoxin: This organism can secrete an en-
terotoxin, very similar to the heat-stable enterotoxin of
Escherichia coli,
that causes diarrhea.
Diagnosis can be made by isolation of this organism
from feces or blood. Treatment does not appear to alter
the course of the gastroenteritis, but patients who have
sepsis should be treated with antibiotics. Although re-
frigeration of food can wipe out many types of bacterial
pathogens,
Yersinia enterocolitica
can survive and grow
in the cold.
Other members of the Enterobacteriaceae family that
you will hear of on the wards include
Edwardsiella, Cit-
robacter, Hafnia,
and
Providencia.
FAMILY VIBRIONACEAE
Vibrio cholera
Figure 9-9
Fig. 9-9. As you can see,
Vibrio cholera
is a curved
gram-negative rod with a single polar flagellum.
Cholera is the diarrheal disease caused by
Vibrio
cholera.
The bacteria are transmitted by the fecal-oral
route, and fecally contaminated water is usually the
culprit. Adults in the U.S., especially travelers, and
children in endemic areas are the groups primarily in-
fected (immunity develops in adults in endemic areas).
Recent epidemics have arisen secondary to poor dis-
posal of sewage in many South American countries
(400,000 cases in Latin America in 1991), and 1993
monsoon floods that mixed feces with potable water in
Bangladesh.
The bacteria multiply in the intestine and cause the
same disease as ETEC, but more severe. As with ETEC,
CHAPTER 9. THE ENTERICS
62
there is no epithelial cell invasion. The bacteria attach
to the epithelial cells and release the cholera toxin,
which is called choleragen. The disease presents with
the abrupt onset of a watery diarrhea (classically de-
scribed as looking like rice water) with the loss of up
to 1 liter of fluid per hour in severe cases. Shock from
isotonic fluid loss will occur if the patient is not rehy-
drated. Like ETEC:
Cholera causes death by dehydration.
Physical findings such as diminished pulses, sunken
eyes, and poor skin turgor will develop with severe de-
hydration.
Choleragen
This toxin has the same mechanism of action as
Es-
cherichia coli's
LT toxin (although choleragen is coded
on the chromosome, while LT is transmitted via a plas-
mid). There is one A subunit (Action) attached to five B
subunits (Binding). The B subunit binds to the GM1
ganglioside on the intestinal epithelial cell surface, al-
lowing entry of the A subunit. In the cell, the A subunit
activates G-protein, which in turn stimulates the activ-
ity of a membrane-bound adenylate cyclase, resulting in
the production of cAMP. Intracellular cAMP results in
active secretion of Na and Cl as well as the inhibition of
Na and Cl reabsorption. Fluid, bicarbonate, and potas-
sium are lost with the osmotic pull of the NaCl as it trav-
els down the intestine.
Microscopic exam of the stool should not reveal
leukocytes (white cells) but may reveal numerous
curved rods with fast darting movements. Treatment
with fluid and electrolytes is lifesaving, and doxycycline
will shorten the duration of the illness.
Vibrio parahaemolyticus
This organism is a marine bacterium that causes gas-
troenteritis after ingestion of uncooked seafood (sushi).
This organism is the leading cause of diarrhea in Japan.
Campylobacter jejuni
(Camping bacteria in the jejunum with nothing bet-
ter to do than cause diarrhea!)
This critter is important!!! This gram-negative rod
that looks like
Vibrio cholera
(curved with a single po-
lar flagellum) is often lost deep in textbooks. Don't let
this happen.
Campylobacter jejuni,
ETEC, and the Ro-
tavirus are the three most common causes of diarrhea
in the world. Estimates are that
Campylobacter jejuni
causes up to 2 million cases of diarrhea a year in the
U.S. alone.

This is a zoonotic disease, like most
Salmonella
(except
Salmonella typhi),
with reservoirs of
Campylobacter je-
juni
in wild and domestic animals and in poultry. The fe-
caloral route via contaminated water is often the mode
of transmission. This organism can also be acquired by
drinking unpasteurized milk. As with most diarrheal ill-
ness, children are the most commonly affected worldwide.
The illness begins with a prodrome of fever and
headache, followed after half a day by abdominal cramps
and a bloody, loose diarrhea. This organism invades the
lining of the small intestine and spreads systemically as
do
Salmonella typhi
and
Yersinia enterocolitica. Campy-
lobacterjejuni
also secretes an LT toxin similar to that
of
Escherichia coli
and an unknown cytotoxin that de-
stroys mucosal cells.
Helicobacter pylori
(formerly called
Campylobacter pylori)
This organism is the most common cause of duo-
denal ulcers and chronic gastritis (inflamed stomach).
(Aspirin products rank second.) It is the second leading
cause of gastric (stomach) ulcers, behind aspirin prod-
ucts. The evidence for this is as follows:
1) Helicobacter pylori
can be cultured from ulcer
craters.
2) Feeding human volunteers
Helicobacter pylori
causes ulcer formation and gastritis.
3) Pepto-Bismol, used for years for gastritis, has bis-
muth salts, which inhibit the growth
of Helicobacter
pylori.
4) Antibiotics help treat duodenal and gastric ulcer
disease: Multiple recent studies have shown that treat-
ment with combinations of bismuth salts, Metronida-
zole, ampicillin, and/or tetracycline, clears
Helicobacter
pylori
and results in a dramatic decrease in both duo-
denal and gastric ulcer recurrence (Veldhuyzen van
Zanten, 1994; Ransohoff, 1994; Sung, 1995).
Fig. 9-10.
Helicobacter pylori
causes duodenal and
gastric ulcers and gastritis. Visualize a Helicopter-
bacteria lifting the cap off a duodenal and gastric ulcer
crater. If you have a more violent disposition, visualize
an Apache helicopter-bacteria shooting hellfire mis-
siles at the stomach.
FAMILY PSEUDOMONADACEAE
Pseudomonas aeruginosa
You are going to hear so much about this bug while
working in the hospital that you will wish the Lord had
never conjured it up. There are two reasons why it is so
important:
1) It colonizes and infects sick, immunocompromised
hospitalized patients, the kind of patient you will take
care of in the hospital.
CHAPTER 9. THE ENTERICS
63
2) The rascal is resistant to almost every antibiotic,
so it has become an art to think up "anti-pseudomonal
coverage." Drug salesmen will always mention the cov-
erage for
Pseudomonas.
Pseudomonas aeruginosa is
an obligate aerobic (non-
lactose fermenter), gram-negative rod. It produces a
green fluorescent pigment (fluorescein) and a blue pig-
ment (pyocyanin), which gives colonies and infected
wound dressings a greenish-blue coloration. It also pro-
duces a sweet grape-like scent, so wound dressings and
agar plates are often sniffed for organism identification.
Pseudomonas aeruginosa
has weak invasive ability.
Healthy people just don't get infections with this guy!
However, once inside a weakened patient, the story
changes. It elaborates numerous exotoxins including
exotoxin A, which has the same mechanism of action
as diphtheria toxin (stops protein synthesis) but is not
antigenically identical. Some strains also possess a cap-
sule that is antiphagocytic and aids in adhesion to tar-
get cells (in the lungs for example).
Important
Pseudomonas
aeruginosa
Infections
1) Pneumonia (see Fig. 16-5)
a) Most cystic fibrosis patients have their lungs
colonized with
Pseudomonas aeruginosa.
These pa-
tients develop a chronic pneumonia, which progres-
sively destroys their lungs.
b) Immunocompromised patients (cancer pa-
tients and intensive care unit patients) are highly
susceptible to pneumonia caused by
Pseudomonas
aeruginosa.
2) Osteomyelitis
a) Diabetic patients have an increased risk of de-
veloping foot ulcers infected with
Pseudomonas
aeruginosa.
The infection can penetrate into the
bone resulting in osteomyelitis.
b) Intravenous (IV) drug abusers have an in-
creased risk of osteomyelitis of the vertebrae or
clavicle.
c) Children develop osteomyelitis secondary to
puncture wounds to the foot.
3) Burn-wound infections: This organism sets up
significant infections of burn wounds, which eventually
lead to a fatal sepsis.
4) Sepsis:
Pseudomonas
sepsis carries an extremely
high mortality rate.
5) Urinary tract infections, pyelonephritis: This
occurs in debilitated patients in nursing homes and in
hospitals. They often have urethral Foley catheters,
which serve as a source of infection.
6) Endocarditis:
Staphylococcus aureus
and
Pseudo-
monas aeruginosa
are frequent causes of right heart
valve endocarditis in IV drug abusers.
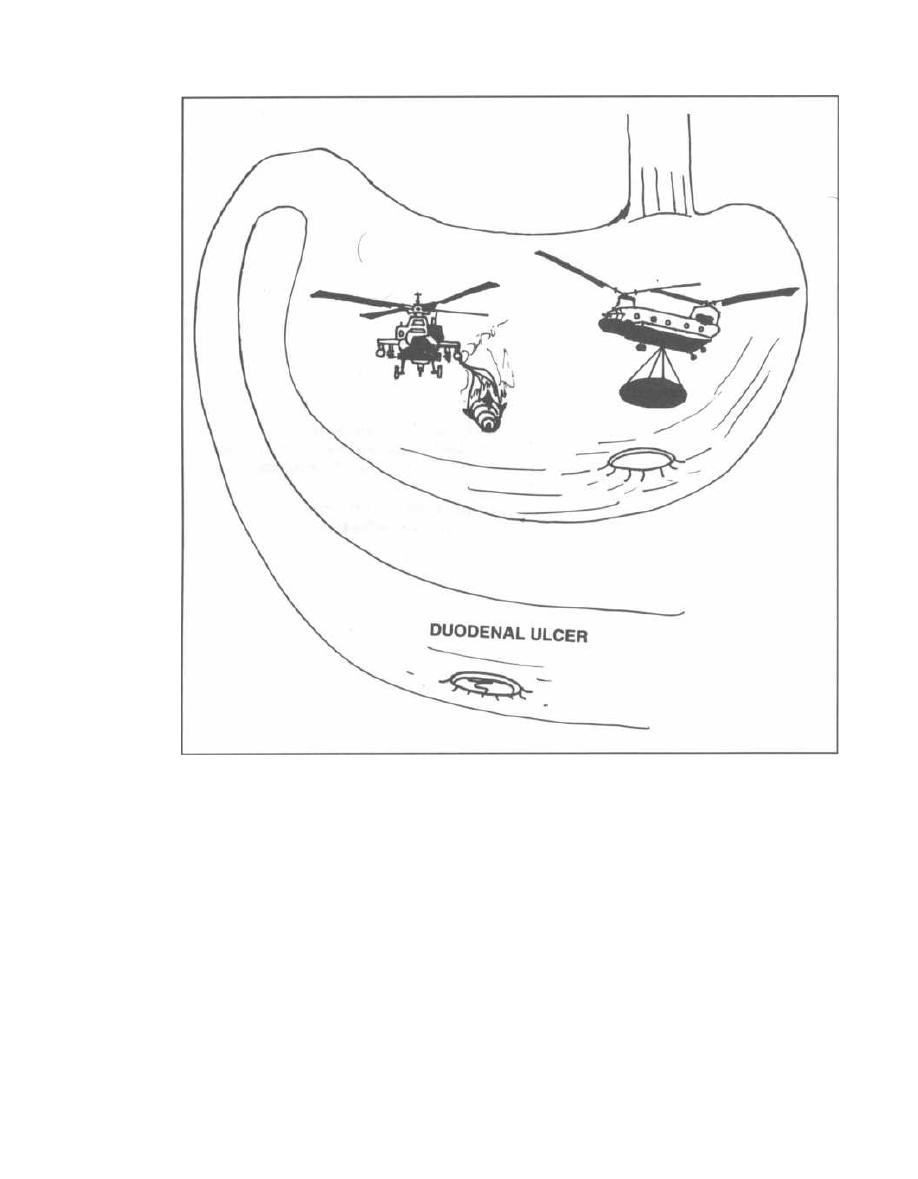
Figure 9-10
7) Malignant external otitis:
A
Pseudomonas
ex-
ternal ear canal infection burrows into the mastoid
bone, primarily in elderly diabetic patients.
8) Corneal infections: This can occur in contact
lens wearers.
Treatment of
Pseudomonas
is complicated as it is re-
sistant to many antibiotics. Chapter 16, Fig. 16-14, lists
all the antibiotics used to treat
Pseudomonas.
An anti-
pseudomonal penicillin is usually combined with an
aminoglycoside for synergy (for example, piperacillin
and gentamicin).
Pseudomonas cepacia
is
rapidly becoming an im-
portant pathogen, infecting hospitalized patients (burn
and cystic fibrosis patients) in a similar manner.
CHAPTER 9. THE ENTERICS
64
FAMILY BACTEROIDACEAE
We have spent so much time studying all the preced-
ing enteric bacteria that you may be surprised to find
out that 99% of the flora of our intestinal tract is made
up of obligate anaerobic gram-negative rods comprising
the family Bacteroidaceae. The mouth and vagina are
also home to these critters.
Bacteroides fragilis
This bacterium is notable for being one of the few
gram-negative bacteria that does not contain lipid A in
its outer cell membrane (NO endotoxin!). However, it
does possess a capsule.

You will become very familiar with Bacteroides frag-
ilis
while studying surgery. This bacterium has low vir-
ulence and normally lives in peace in the intestine.
However, when a bullet tears into the intestine, when a
seat belt lacerates the intestine in a car wreck, when ab-
dominal surgery is performed with bowel penetration,
or when the intestine ruptures secondary to infection
(appendicitis) or ischemia, THEN the bacteria go wild in
the peritoneal cavity, forming abscesses. An abscess is
a contained collection of bacteria, white cells, and dead
tissue. Fever and sometimes systemic spread accom-
pany the infection.
'Phis abscess formation is also seen in obstetric and
gynecologic patients. Abscesses may arise in a patient
with a septic abortion, pelvic inflammatory disease
(tubo-ovarian abscess), or an intrauterine device (IUD)
for birth control.
Bacteroides fragilis is rarely present in the mouth, so
it is rarely involved in aspiration pneumonias.
Following abdominal surgery, antibiotics that cover
anaerobes are given as prophylaxis against Bacteroides
fragilis. These include clindamycin, metronidazole
( Flagyl), chloramphenicol, and others (see Chapter 16,
Fig. 16-15). If an abscess forms, it must be surgically
drained.
Bacteroides
melaninogenicus
This organism produces a black pigment when grown
on blood agar. Hence, the name melaninogenicus. It
lives in the mouth, vagina, and intestine, and is usually
involved in necrotizing anaerobic pneumonias caused
by aspiration of lots of sputum from the mouth (during
a seizure or drunken state). It also causes periodontal
disease.
CHAPTER 9. THE ENTERICS
65
Fusobacterium
This bacterium is just like Bacteroides melaninogeni-
cus in that it also causes periodontal disease and aspi-
ration pneumonias. Fusobacterium can also cause
abdominal and pelvic abscesses and otitis media.
ANAEROBIC GRAM-POSITIVE COCCI
Peptostreptococcus (strip or chain of cocci) and Pepto-
coccus
(cluster of cocci) are gram-positive anaerobes
that are part of the normal flora of the mouth, vagina,
and intestine. They are mixed with the preceding or-
ganisms in abscesses and aspiration pneumonias.
Members of the Streptococcus viridans group, dis-
cussed in Chapter 4, are mentioned here because they
are gram-positive, microaerophilic, and are frequently
isolated from abscesses (usually mixed with other anaer-
obic bacteria). These oxygen-hating critters have many
names (such as Streptococcus anginosus and Streptococ-
cus milleri) and are a part of the normal GI flora.
Fig. 9-11.
Summary of enteric bacteria.
References
Veldhuyzen van Zanten SF, Sherman PM.
Helicobacter pylori
infection as a cause of gastritis, duodenal ulcer, gastric can-
cer and nonulcer dyspepsia: a systematic overview. Can
Med Assoc F
1994;150:177-185.
Veldhuyzen van Zanten SF, Sherman PM. Indications for
treatment of
Helicobacter pylori
infection: a systematic
overview. Can Med Assoc F
1994;150:189-198.
Ransohoff DF. Commentary. ACP Journal Club
1994; 120:
62-63.
Sung, JY, et al. Antibacterial treatment of gastric ulcers asso-
ciated with
Helicobacter pylori.
New Engl J Med
1995;332:
139-42.
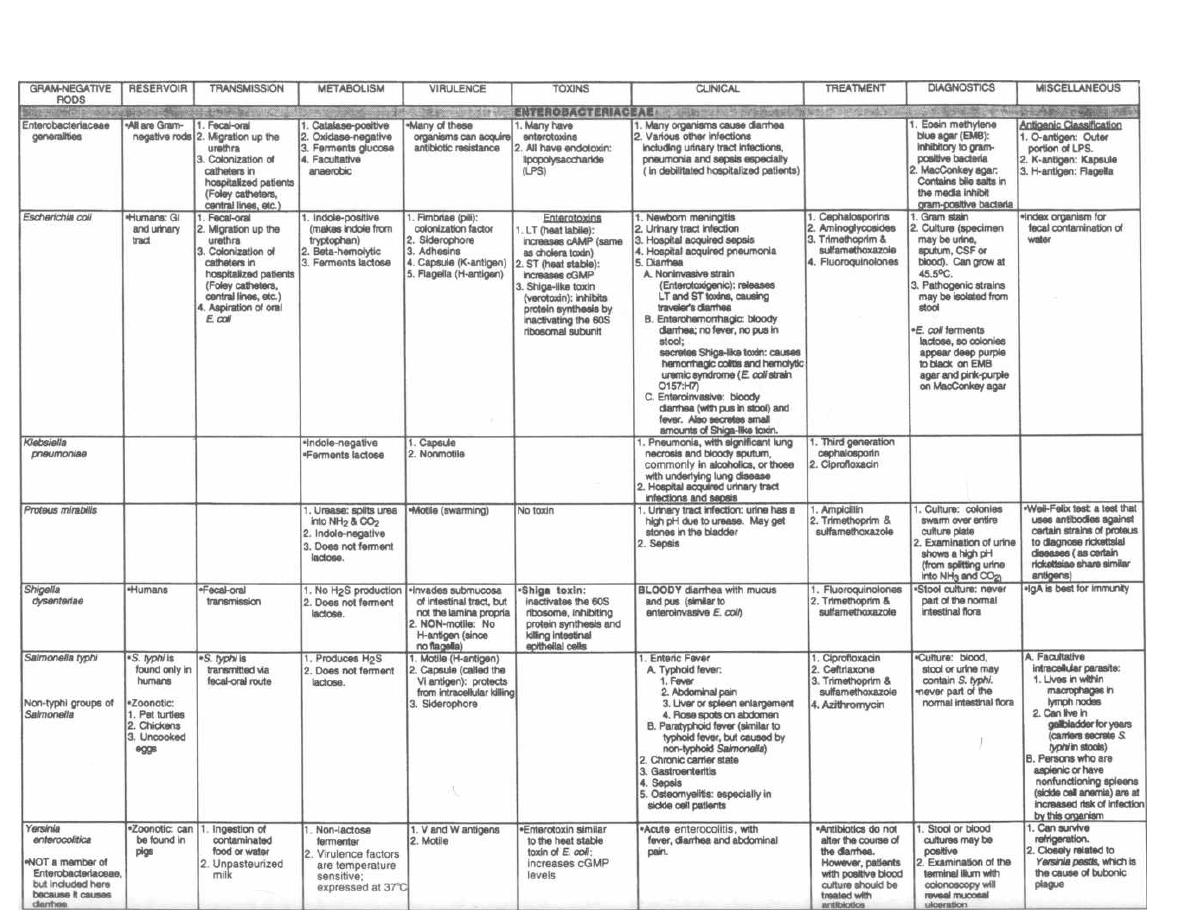
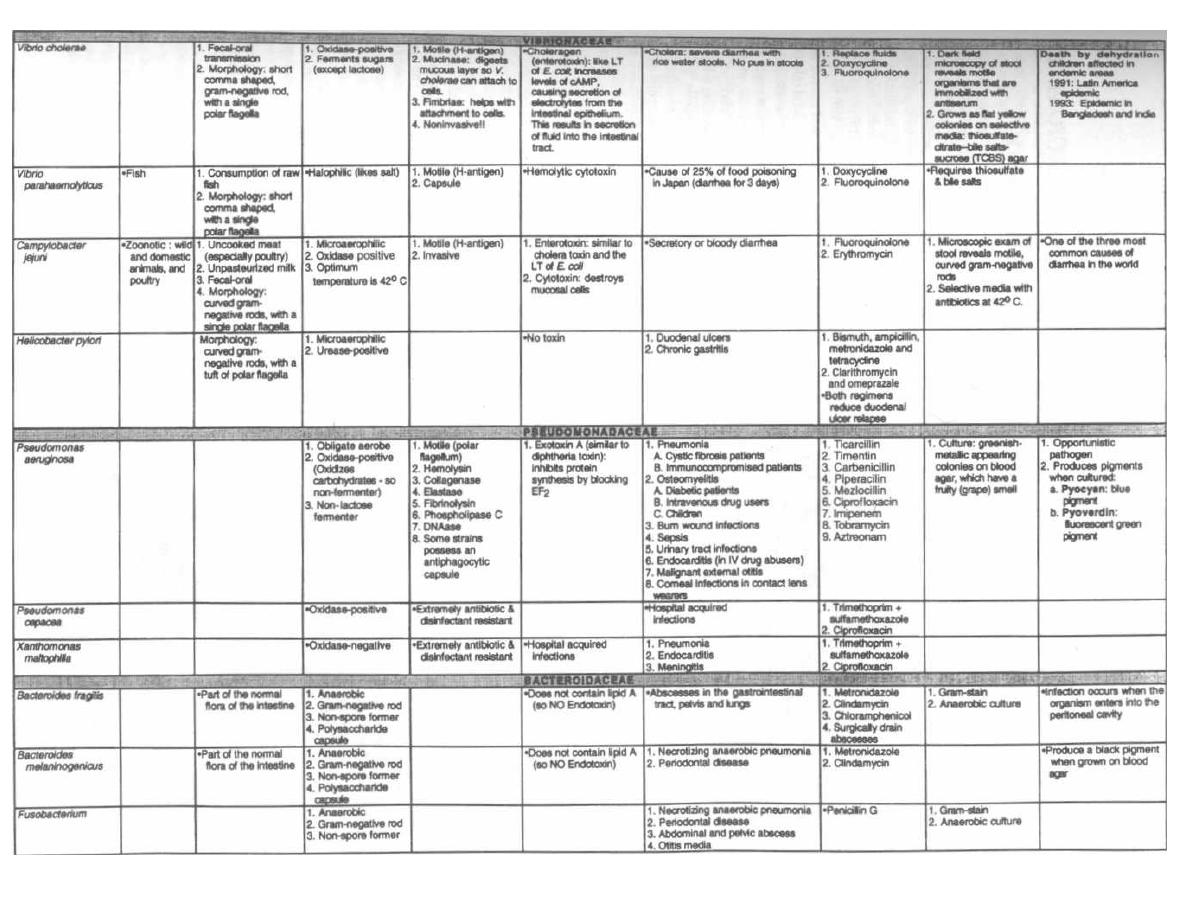
Figure 9-11 (continued)
M. Gladwin and B. Trattler,
Clinical
Microbiology Made Ridiculously Simple ©MedMaster

CHAPTER 10.
HAEMOPHILUS, BORDETELLA,
and
LEGIONELLA
The gram-negative rods
Haemophilus influenzae, Borde-
tella pertussis
and
Legionella pneumophila
are grouped
together because they are all acquired through the respi-
ratory tract. This makes sense if you consider the species
names:
influenzae
(the flu-an upper respiratory ill-
ness),pertussis
(cough), and
pneumophila
(lung loving).
Haemophilus influenzae
The name
Haemophilus influenzae
describes some of
its properties:
Haemophilus
means "blood loving." This organism
requires a blood-containing medium for growth. Hema-
tin found in blood is necessary for the bacterium's cy-
tochrome system. Blood also contains NAD', needed for
metabolic activity.
influenzae:
This bacterium often attacks the lungs
of persons debilitated by a viral influenza infection.
During the 1890 and 1918 influenza pandemics, scien-
tists cultured
Haemophilus influenzae
from the upper
respiratory tracts of "flu" patients, leading them to in-
correctly conclude
that Haemophilus influenzae
was the
etiologic agent of the flu.
Haemophilus influenzae is
an obligate human para-
site that is transmitted via the respiratory route. Two
important concepts help us understand how this critter
causes disease:
1) A polysaccharide capsule confers virulence: There
are 6 types of capsules, designated a, b, c, d, e, and f. Of
these, type b is commonly associated with invasive
Haemophilus influenzae
disease in children, such as
meningitis, epiglottitis, and septic arthritis.
Capsule b = bad
Nonencapsulated strains of
Haemophilus influenzae
can colonize the upper respiratory tract of children and
adults. They lack the virulent invasiveness of their en-
capsulated cousins and can only cause local infection.
They frequently cause otitis media in children as well as
respiratory disease in adults weakened by preexisting
lung disease, such as chronic bronchitis from smoking
or recent viral influenza infection.
2) Antibodies to the capsule are lacking in infants
and children between 6 months and 3 years of age. The
mother possesses antibodies against the b capsule
which she has acquired in her lifetime. She passes these
antibodies to the fetus transplacentally and in her
breast milk. These "passively" acquired antibodies last
for about 6 months. It takes 3-5 years of
Haemophilus
influenzae'
colonization and infection for children to de-
velop their own antibodies. So there is a window during
which children are sitting ducks for the invasive
Haemophilus influenzae.
68
Haemophilus influenzae type b
1) Meningitis: This is the most serious infection
caused by encapsulated
Haemophilus influenzae
type b.
Prior to the introduction of vaccination of U.S. children
in 1991, it was the main cause of meningitis in young
children between the age of 6 months to 3 years (more
than 10,000 cases per year). Following inhalation, this
organism invades the local lymph nodes and blood-
stream, and then penetrates into the meninges. Since
infants usually do not display the classic stiff neck, non-
specific signs such as fever, vomiting, and altered men-
tal status are the clues to this potentially fatal infection.
Although mortality with appropriate antibiotics is
less than 5%, up to half of infected children will still
have permanent residual neurologic deficits, such as
mental retardation, seizures, language delay, or deaf-
ness. When a bacterial meningitis is treated with an-
tibiotics, the killed bacteria lyse and release cellular
antigens, such as LPS lipid A (endotoxin), resulting in a
violent immune response that destroys neurons as well
as bacteria. Recent studies show that treatment with
steroids 15-20 minutes before giving N antibiotics will
decrease this risk of developing neurologic deficits. It is
theorized that the steroids limit the inflammatory re-
sponse to the dead bacteria's antigens while allowing
bacterial killing.
2) Acute epiglottitis:
Haemophilus influenzae
type
b can also cause rapid swelling of the epiglottis, ob-
structing the respiratory tract and esophagus. Follow-
ing a sore throat and fever, the child develops severe
upper airway wheezing (stridor) and is unable to swal-
low. Excessive saliva will drool out of the child's mouth
as it is unable to pass the swollen epiglottis. The large,
red epiglottis looks like a red cherry at the base of the
tongue. If you suspect this infection, do not examine the
larynx unless you are ready to insert an endotracheal
breathing tube because manipulation can cause laryn-
geal spasm. This may cause complete airway obstruc-
tion that can only be bypassed with a tracheotomy.
3) Septic arthritis:
Haemophilus influenzae
type b
is the most common cause of septic arthritis in infants.
Most commonly, a single joint is infected, resulting in
fever, pain, swelling and decreased mobility of the joint.
Examination of the synovial fluid (joint fluid) by Gram
stain reveals the pleomorphic gram-negative rods.
4) Sepsis: Children between 6 months to 3 years pre-
sent with fever, lethargy, loss of appetite, and no evi-
dence of localized disease (otitis media, meningitis, or
epiglottitis). Presumably the bacteria invade the blood-
stream via the upper respiratory tract. Since the spleen
is the most important organ in fighting off infection by
encapsulated bacteria, it is not surprising that children

CHAPTER 10. HAEMOPHILUS, BORDETELLA, AND LEGIONELLA
with absent or non-functioning spleens (either by
surgery or with sickle-cell disease) are at highest risk.
Prompt identification and treatment will prevent
Haemophilus influenzae type b from invading the
meninges, epiglottis, or a joint.
Meningitis, epiglottitis, and bacterial sepsis are
rapidly fatal without antibiotic therapy. Ampicillin
used to be the drug of choice prior to the development of
resistance. Ampicillin resistance is transmitted by a
plasmid from strain to strain of Haemophilus influen-
zae. Currently, a third generation cephalosporin, such
as cefotaxime or ceftriaxone, is the drug of choice for
serious infections. Ampicillin or amoxicillin can be
used for less serious infections, such as otitis media.
Vaccination
( Hib capsule vaccine)
The key to controlling this organism is to stimulate
the early generation of protective antibodies in young
children. However, it is difficult to stimulate antibody
formation in the very young.
The first vaccine, consisting of purified type b cap-
sule, was effective only in generating antibodies in chil-
dren older than 18 months. A second new vaccine is
composed of the Haemophilus influenzae type b (Hib)
capsule and diphtheria toxin. The addition of the diph-
theria toxin activates T-lymphocytes and antibodies
against the b capsule. Vaccination with the Hib capsule
of children in the U.S. at ages 2, 4, 6, and 15 months
(along with the DTP and polio vaccines) has dramati-
cally reduced the incidence of Haemophilus influenzae
i nfection. Acute Haemophilus influenzae epiglottitis is
now rarely seen in U.S. emergency rooms.
Hib, Hib, Hurray!
Other efforts involve immunizing women in the
eighth month of pregnancy, resulting in increased anti-
body secretion in breast milk (passive immunization).
Haemophilus ducreyi
This species is responsible for the sexually transmitted
disease chancroid. Clinically, patients present with a
painful genital ulcer. Unilateral painful swollen in-
guinal lymph nodes rapidly develop in half of infected
persons. The lymph nodes become matted and will rup-
ture, releasing pus.
The differential diagnosis includes:
1) Syphilis ( Treponema pallidum): It is extremely
i mportant to exclude syphilis as the cause of the ulcer.
Remember that the ulcer of syphilis is painless and the
associated adenopathy is bilateral, painless, and non-
suppurative (no pus).
6 9
2) Herpes ( Herpes simplex virus 1 and 2): Herpetic
lesions start as vesicles (blisters), yet once they break
they can be misdiagnosed as chancroid, especially be-
cause they are painful. Herpes is usually accompanied
by systemic symptoms such as myalgias and fevers.
Chancroid does not usually produce systemic symptoms.
3) Lymphogranuloma venereum ( Chlamydia tra-
chomatis): LGV has painless matted suppurative in-
guinal lymph nodes that develop much more slowly
than chancroid. The primary ulcer of LGV disappears
before the nodes enlarge, whereas with chancroid they
coexist.
Treat chancroid with erythromycin or trimetho-
prim/sulfamethoxazole. Effective treatment of geni-
tal ulcers is crucial, because these open lesions create a
break in the skin barrier, increasing the risk of HIV
transmission.
Gardnerella vaginalis
(formerly Haemophilus vaginalis)
This organism causes bacterial vaginitis in conjunc-
tion with anaerobic vaginal bacteria. Women with
vaginitis develop burning or pruritis (itching) of the
labia, burning on urination (dysuria), and a copious,
foul-smelling vaginal discharge that has a fishy odor. It
can be differentiated from other causes of vaginitis
(such as Candida or Trichomonas) by examining a slide
of the vaginal discharge (collected from the vagina dur-
ing speculum exam) for the presence of clue cells. Clue
cells are vaginal epithelial cells that contain tiny pleo-
morphic bacilli within the cytoplasm.
Treat this infection with metronidazole, which cov-
ers Gardnerella as well as co-infecting anaerobes. As a
note, this species was separated from the genus
Haemophilus because it does not require X-factor or V-
factor for growth in culture.
Bordetella pertussis
This bacterium is named:
Bordetella
because it was discovered in the early
1900's by two scientists named Bordet and Gengou. It
seems that Bordet got the better end of the deal!
Pertussis
means "violent cough." Bordetella pertus-
sis causes whooping cough.
Exotoxin Weapons
Bordetella pertussis is a violently militant critter
with a (gram) negative attitude. He is a gram-negative
rod armed to the hilt with 4 major weapons (virulence
factors). These virulence factors allow him to attach to
the ciliated epithelial cells of the trachea and bronchi.
He evades the host's defenses and destroys the ciliated
cells, causing whooping cough.

CHAPTER 10. HAEMOPHILUS, BORDETELLA, AND LEGIONELLA
1) Pertussis toxin: Like many bacterial exotoxins
this toxin has a B subunit that Binds to target cell re-
ceptors, "unlocks" the cell, allowing entry of the A sub-
unit.
The
A subunit (A for Action) activates
cell-membrane-bound G regulatory proteins, which in
turn activate adenylate cyclase. This results in an out-
pouring of cAMP, which activates protein kinase and
other intracellular messengers. The exact role of this
toxin in whooping cough is not entirely clear, but it has
3 observed effects: a) histamine sensitization, b) in-
crease in insulin synthesis, and c) promotion of lympho-
cyte production and inhibition of phagocytosis.
2)
Extra cytoplasmic adenylate cyclase: When
attacking the bronchi, Bordetella pertussis throws its
adenylate cyclase grenades. They are swallowed by host
neutrophils, lymphocytes, and monocytes. The internal-
ized adenylate cyclase then synthesizes the messenger
cAMP, resulting in impaired chemotaxis and impaired
generation of
H2O2
and superoxide. This weakens the
host defense cells' ability to phagocytose and clear the
bacteria.
3) Filamentous hemagglutinin (FHA): Bordetella
pertussis does not actually invade the body. It attaches
to ciliated epithelial cells of the bronchi and then re-
leases its damaging exotoxins. The FHA, a pili rod ex-
tending from its surface, is involved in this binding.
Antibodies directed against the FHA prevent binding
and disease, and thus they are protective.
4) Tracheal cytotoxin: This toxin destroys the cili-
ated epithelial cells, resulting in impaired clearance of
bacteria, mucus, and inflammatory exudate. This toxin
is probably responsible for the violent cough.
Whooping Cough
The number of cases of whooping cough has decreased
dramatically since vaccination programs began. In the
prevaccination era in the United States, there were ap-
proximately 100-300 thousand cases a year, and now
only 1-4 thousand!!! Prior to the development of the vac-
cine, children between the ages of
1-5
were most likely
to catch this disease.
The majority of cases today occur in unimmunized in-
fants younger than 1 year. Infants younger than 6
months used to be protected by maternal antibodies
that crossed the placenta during pregnancy. However,
the vaccine only provides a high level of protective anti-
bodies during the first
15
years of life, so most mothers
do not have protective antibodies to pass to their in-
fants. Therefore, unimmunized infants under 1 year are
very susceptible to this infection today. Since the vac-
cine only provides immunity for approximately
15
years, young adults are another group that is currently
at a higher risk for acquiring whooping cough.
Whooping cough is a highly contagious disease with
transmission occurring via respiratory secretions on the
7 0
hands or in an aerosolized form. A week-long incubation
period is followed by 3 stages of the disease:
1) Catarrhal stage: This stage lasts from
1-2
weeks
and is similar to an upper respiratory tract infection, with
low-grade fevers, runny nose, sneezing, and mild cough.
It is during this period that the disease is most contagious.
2)
Paroxysmal stage: The fever subsides and the
i nfected individual develops characteristic bursts of
nonproductive cough. There may be
15-25
of these at-
tacks per day, and the person may appear normal be-
tween events. The attacks consist of
5-20
forceful
coughs followed by an inspiratory gasp through the nar-
rowed glottis. This inspiration sounds like a whoop.
During these paroxysms of coughing the patient can be-
come hypoxemic and cyanotic (blue from low oxygen),
the tongue may protrude, eyes bulge, and neck veins en-
gorge. Vomiting often follows an attack. The paroxys-
mal stage can last a month or longer. The illness is more
severe in the young, with up to
75%
of infants less than
6 months of age and 40% of infants and young children
more than 6 months requiring hospitalization.
Infants and partially immunized (wearing off) chil-
dren and adults may not have the typical whoop. In-
fants can have cough and apnea spells (no breathing).
Adults may present with a persistent cough.
Examination of the white blood cells will surprisingly
reveal an increase in the lymphocyte count with just a
modest increase in the neutrophils (more like a viral pic-
ture). The increased number of lymphocytes seems to be
one of the manifestations of the pertussis toxin.
3) Convalescent stage: The attacks become less
frequent over a month, and the patient is no longer con-
tagious.
Since this organism will not grow on cotton, speci-
mens for culture are collected from the posterior phar-
ynx with a calcium alginate swab. This swab is
i nserted into the posterior nares and the patient is then
instructed to cough. The swab is then wiped on a special
culture medium with potato, blood, and glycerol agar,
called the Bordet-Gengou medium. At most hospi-
tals, identification of this bacterium can be made with
rapid serological tests (ELISA).
Treatment is primarily supportive. Infants are hospi-
talized to provide oxygen, suctioning of respiratory secre-
tions, respiratory isolation, and observation. Treatment
of infected individuals with erythromycin in the pro-
dromal or catarrhal stage may prevent the disease. Later
therapy during the paroxysmal stage does not alter the
course of illness but may decrease bacterial shedding.
Household contacts should receive erythromycin also.
Vaccination
The vaccine currently used in the U.S. consists of
heat-killed organisms and includes the pertussis toxin,

CHAPTER 10. HAEMOPHILUS, BORDETELLA, AND LEGIONELLA
FHA, and adenylate cyclase. It is combined with the for-
malin inactivated tetanus and diphtheria toxoids to
form the DPT (Diphtheria-Pertussis-Tetanus) vaccine,
and is given at 2, 4, 6, and 15-18 months of age. This
vaccine has been very effective in reducing the number
of whooping cough cases but carries a price. Infants may
develop side effects such as local swelling and pain and
systemic fever, persistent crying, and, rarely, limpness
(hypotonicity) and seizures.
In efforts to reduce these adverse effects new vaccines
have been developed that are composed only of inacti-
vated proteins such as pertussis toxin, FHA, and others
(such as pertactin and fimbrial antigens). In two recent
large studies, these vaccines were found to be safer and
worked better than the U.S. whole-cell vaccine!!! (Greco,
1996; Gustafsson, 1996).
Legionella pneumophila
( Legionnaires' Pneumonia)
Legionella pneumophila is
an aerobic gram-negative
rod that is famous for causing an outbreak of pneumo-
nia at an American Legion convention in Philadelphia
i n 1976 (thus its name).
This organism is ubiquitous in natural and man-
made water environments. Aerosolized contaminated
water is inhaled, resulting in infection. Sources that
have been identified during outbreaks have included air
conditioning systems, cooling towers, and whirlpools.
Outbreaks have even been associated with organism
growth in shower heads and produce mist machines in
supermarkets!!! Person-to-person transmission has not
been demonstrated.
Like
Mycobacterium tuberculosis,
this organism is a
facultative intracellular parasite that settles in the
lower respiratory tract and is gobbled up by macro-
phages. This means that once it has been phagocytosed,
it inhibits phagosome-lysosome fusion, surviving and
replicating intracellularly.
Legionella is
responsible for diseases ranging from
asymptomatic infection and a flulike illness called Pon-
7 1
tiac fever to a severe pneumonia called Legionnaires'
disease:
1) Pontiac fever: Like influenza, this disease in-
volves headache, muscle aches, and fatigue, followed
by fever and chills. Pontiac fever strikes suddenly and
completely resolves in less than one week. Pontiac
fever was so-named for the illness that struck 95% of
the employees of the Pontiac, Michigan, County
Health Department. The causative agent was identi-
fied as
Legionella pneumophila
carried by the air con-
ditioning system.
2) Legionnaires' disease: Patients develop very
high fevers and a severe pneumonia.
Legionella pneumophila is
one of the most common
causes of community acquired pneumonia and is esti-
mated to be diagnosed correctly in only 3% of cases! It
should be suspected in all patients who have pneumo-
nia who are over 50 years of age and especially if they
are smokers or if the sputum gram stain reveals neu-
trophils and very few organisms.
(Legionella is so
small
it is hard to see on gram stain.)
Treat with erythromycin because this organism has
a beta-lactamase making it resistant to penicillins.
Then attempt to determine the source of
Legionella. Is
the air conditioning system contaminated?
Fig. 10-1. Summary of
Haemophilus, Bordetella
and
Legionella.
References
Greco D, Salmaso S, et al. A controlled trial of two acellular
vaccines and one whole-cell vaccine against pertussis. N.
Eng. J. Med. 1996;334:341-8.
Gustafsson L, Hollander H0, et al. A controlled trial of a two-
component acellular, a five-component acellular, and a
whole-cell pertussis vaccine. N. Eng. J. Med. 1996;334:349-
55.
Hewlett EL. Bordetella species. In: Mandell GL, Bennett JE,
Dolin R. Editors. Principles and Practice of Infectious Dis-
eases. 4th edition. New York: Churchill Livingstone
1995;2078-2084.
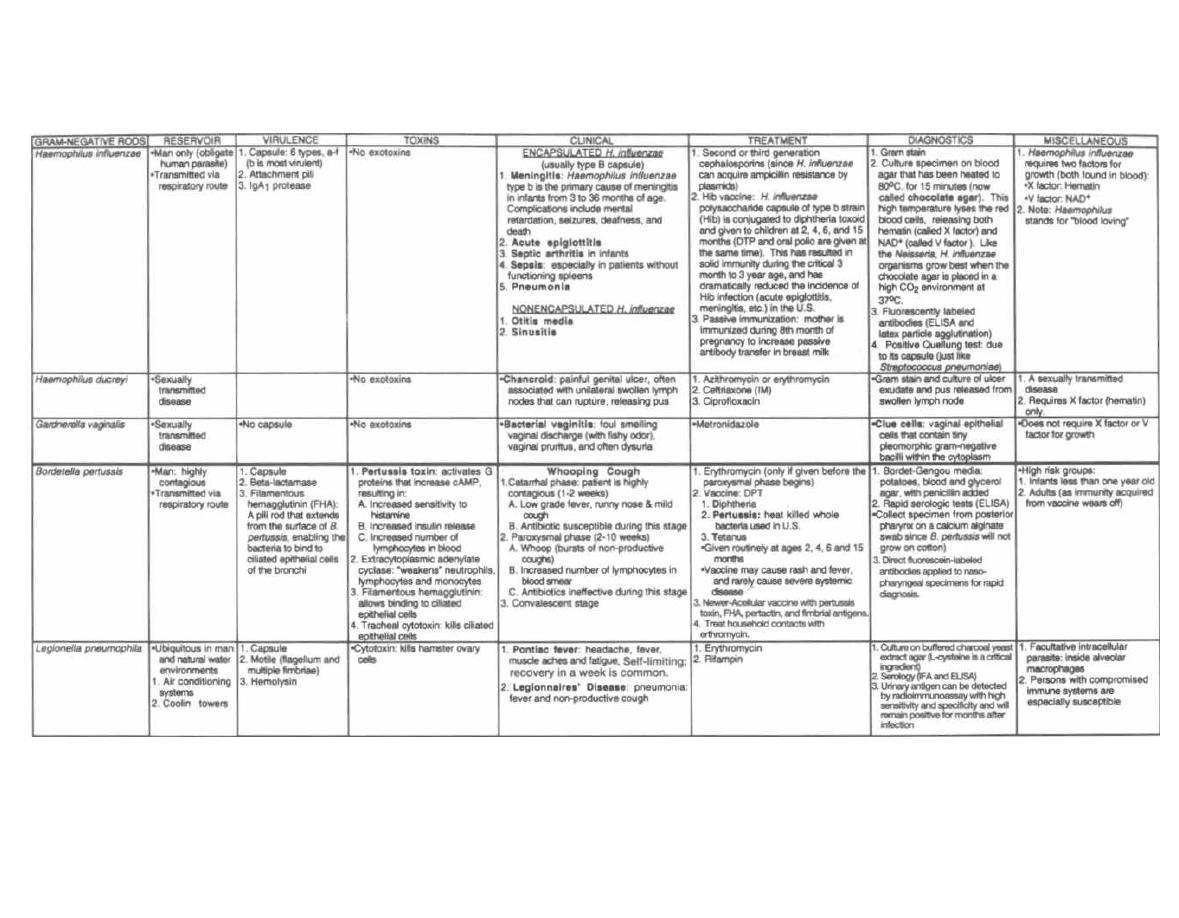
Figure 10-1 HAEMOPHILUS, BORDETELLA AND LEGIONELLA
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple ©MedMaster
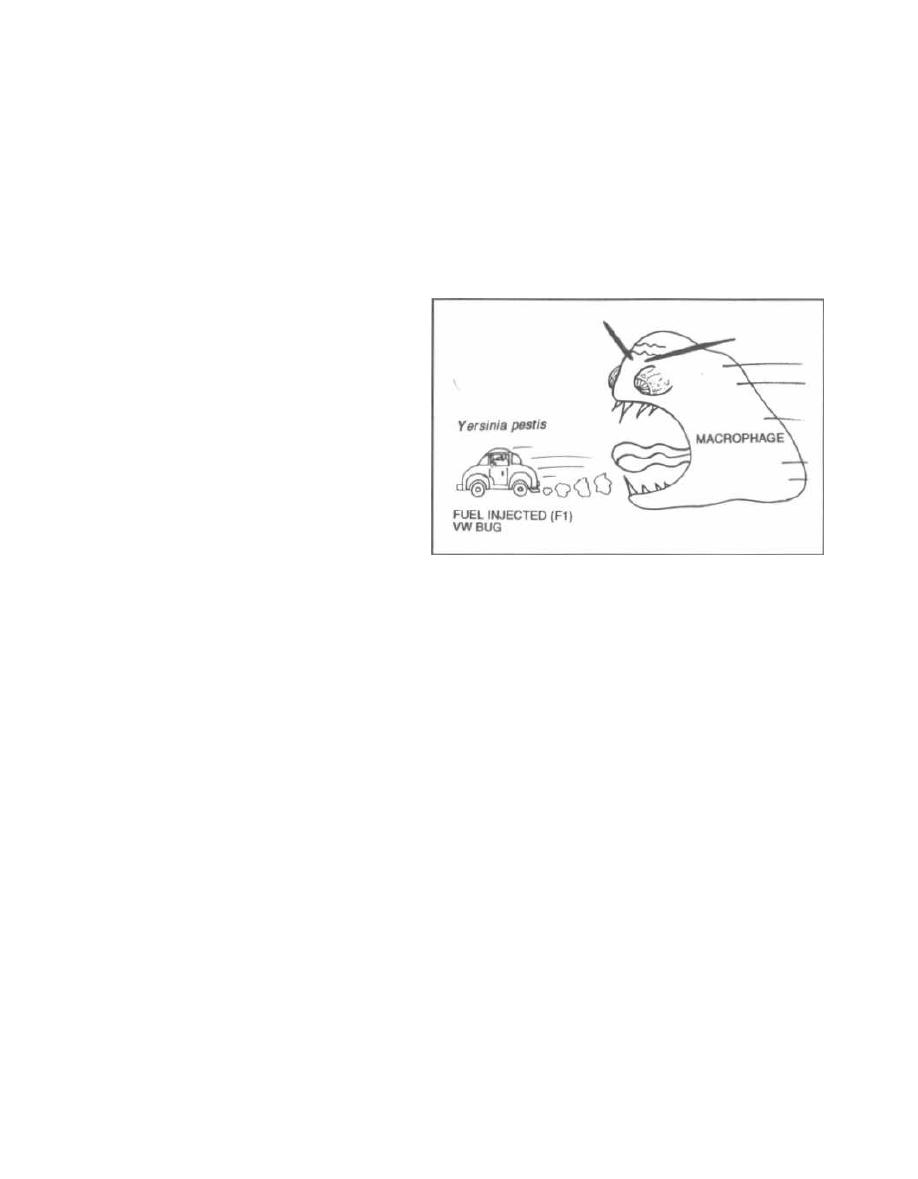
CHAPTER 11. YERSINIA, FRANCISELLA, BRUCELLA,
AND PASTEURELLA
These organisms have been included in the same chap-
ter because they share many characteristics
(Pas-
teurella
only shares the first 2):
1) They are all gram-negative rods (bacilli).
2) All of these are zoonotic diseases (i.e., they are pri-
marily diseases of animals).
3) These bacteria are very virulent and are able to
penetrate any body area they touch. This can occur on
the skin following an insect bite, animal bite, or direct
contact with an animal. This can also occur in the lungs
after inhalation of infected aerosolized matter.
4) From the site of contact (usually the skin) the bac-
teria are phagocytosed by macrophages. They can sur-
vive inside the macrophages and so are facultative
intracellular organisms. They migrate to the re-
gional lymph nodes, set up infection there, and then
move to the bloodstream and other organs, such as the
liver, spleen, and lungs.
Like other facultative intracellular organisms (see
Fig. 2-7) immunity is cell-mediated, and intradermal
injections of bacterial extracts will elicit a delayed-type-
hypersensitivity (DTH) reaction. This reaction results
in skin swelling and induration (hardening) at the in-
jection site 1-2 days later. The presence of swelling in-
dicates previous exposure to the bacteria and can be
used as a diagnostic test (see discussion of DTH and in-
tradermal skin testing in Chapter 14, page 104)
5) The common treatment is an aminoglycoside
(gentamicin or streptomycin) and/or doxycycline,
which must be given for a prolonged period so as to
reach the hidden intracellular bacteria.
Yersinia pestis
(Bubonic Plague)
You have all heard of bubonic plague and that rats
were somehow involved. Rats are the
PESTS (Yersinia
pestis)
that harbor this disease, while fleas serve as
vectors, carrying
Yersinia pestis
to humans. Bubonic
plague destroyed one fourth of the population of Eu-
rope in the 14th century. Later outbreaks moved from
China to India (where the disease killed 10 million)
and in the 1900's to San Francisco. The organism now
resides in squirrels and prairie dogs of the southwest-
ern U. S.
The Fl, V, and W virulence factors enable this or-
ganism to resist destruction after phagocytosis (faculta-
tive intracellular organism):
1) Fraction 1 (Fl): This capsular antigen has an-
tiphagocytic properties.
73
2) V and W antigens: These antigens, which are a
protein and lipoprotein respectively, are unique to the
Yersinia
genus. Their actions are unknown.
Fig. 11-1. Visualize a rat riding in a Fuel Injected
(Fl), VW bug, being pursued by a macrophage. These
three virulence factors are involved in
Yersinia pestis'
resistance to destruction after phagocytosis.
Figure 11-1
Fig.
11-2.
Yersinia pestis
is a gram-negative bac-
terium with a bipolar staining pattern. The ends of the
rod-shaped bacterium take up more stain than the cen-
ter. Three mammals fall prey to
Yersinia pestis:
wild ro-
dents, domestic city rodents, and humans. The bacteria
reside in the wild rodent population between epidemics
and are carried from rodent to rodent by the flea. When
wild rodents come into contact with domestic city rats
(during droughts when wild rodents forage for food),
fleas can then carry the bacteria to domestic rats. As the
domestic rat population dies, the fleas become hungry
and search out humans.
During interepidemic periods (we are in one now),
bubonic plague may be contracted during camping,
hunting or hiking. The human victim either touches a
dead infected rodent or is bitten by an infected flea.
The bacteria invade the skin and are gobbled up by
macrophages. They continue to reproduce intracellu-
larly and within a week move to the nearest lymph
nodes, usually the inguinal nodes
(boubon
is the Greek
word for "groin"). The nodes swell like eggs and become
hot, red, and painful. Fever and headache set in. The
bacilli invade the bloodstream, liver, lungs, and other
organs. Hemorrhages under the skin cause a blackish
discoloration, leading people to call bubonic plague the
"Black Death." Without treatment, death can occur in a
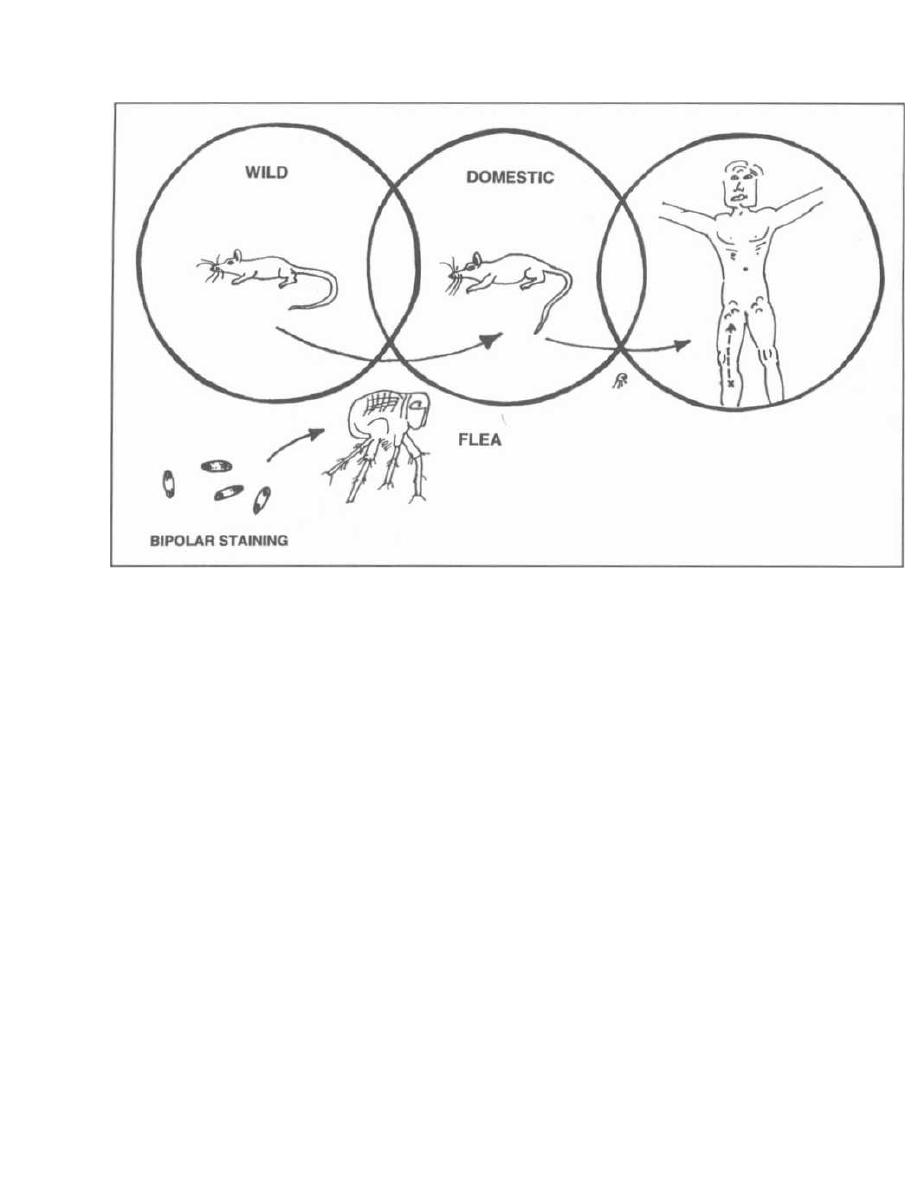
Figure 11-2
few days. During epidemics, the disease can also be seen
as pneumonic plague with pneumonia and human-to-
human transmission by aerosolized bacteria.
If you see a patient who has been camping in Arizona
or New Mexico and has developed fever, have a high in-
dex of suspicion. You may want to start gentamicin
right away. You can't depend on the presence of swollen
lymph nodes: Between 1980 and 1984, 25% of the cases
in New Mexico did not have lymph node involvement.
This disease is deadly if untreated! About 75% of un-
treated people die!
Control of epidemics involves DDT for the fleas and
destruction of the rats. If you only kill the rats, the
starving fleas will feed on humans instead!
Another species of
Yersinia
called
Yersinia enterocol-
itica
infects the colon and is closely related to Es-
cherichia coli
(see Chapter 9 page 61).
Francisella tularensis
(Tularemia)
Tularemia is a disease that resembles bubonic plague
so closely that it is always included in the differential di-
agnosis when considering bubonic plague. This disease
is most commonly acquired from handling infected rab-
bits and from the bites of ticks and deerflies. More
than a hundred creatures carry this bacterium, includ-
CHAPTER 11. YERSINIA, FRANCISELLA, BRUCELLA, AND PASTEURELLA
74
ing rabbits, other mammals, and even reptiles and fish.
Tularemia is distributed all over the U.S.
Fig. 11-3.
Francis ( Francisella)
the rabbit (rabbit
vector) is playing in the
Tulips (Tularensis).
One ear
has a tick, the other a deerfly.
Like
Yersinia pestis,
this organism is extremely viru-
lent and can invade any area of contact, resulting in
more than one disease presentation. The most impor-
tant diseases caused by
Francisella tularensis
are the
ulceroglandular and pneumonic diseases:
1) Ulceroglandular tularemia: Following the bite
of a tick or deerfly, or contact with a wild rabbit, a well-
demarcated hole in the skin with a black base develops.
Fever and systemic symptoms develop, and the local
lymph nodes become swollen, red, and painful (some-
times draining pus). The bacteria can then spread to the
blood and other organs. Note that these symptoms are
almost identical to bubonic plague, but the skin ulcer is
usually absent in the plague and the mortality rate is
not nearly as high as in bubonic plague, reaching 5% for
ulceroglandular tularemia.
2) Pneumonic tularemia: Aerosolization of bacte-
ria during skinning and evisceration of an infected
rabbit or hematogenous spread from the skin (ulcerog-

CHAPTER 11. YERSINIA, FRANCISELLA, BRUCELLA, AND PASTEURELLA
Figure 11-3
landular tularemia) to the lungs can lead to a lung in-
fection (pneumonia).
Francisella tularensis
can also invade other areas of
contact such as the eyes (oculoglandular tularemia)
and the gastrointestinal tract (typhoidal tularemia).
Because this bacterium is so virulent (just 10 organ-
isms can cause disease), most labs will not culture it
from blood or pus. For the same reason it is not advis-
able to drain the infected lymph nodes. Diagnosis rests
on the clinical picture, a skin test similar to the PPD for
tuberculosis, and the measurement of the titers of anti-
bodies to
Francisella tularensis.
Brucella
(Brucellosis)
All the names of
Brucella
species are based on the an-
imal they infect:
•
Brucella melitensis
(goats)
•
Brucella abortus
(causes abortions in cows)
•
Brucella suis (pigs)
•
Brucella canis
(dogs)
75
Humans acquire
Brucella
from direct contact with
infected animal meat or aborted placentas, or inges-
tion of infected milk products. The incidence of this dis-
ease worldwide is greater than that of both bubonic
plague and tularemia. In the U. S., however, it is not
very common because cattle are immunized and milk is
pasteurized.
Fig. 11-4.
If you do see a patient with brucellosis, he
will most likely be a worker in the meat-packing indus-
Figure 11-4

CHAPTER 11. YERSINIA, FRANCISELLA, BRUCELLA, AND PASTEURELLA
try (beef), a veterinarian, a farmer, or a traveler who
consumes dairy (cow or goat) products in Mexico or else-
where.
Like the other bacteria in this chapter,
Brucella
pen-
etrates the skin, conjunctiva, lungs, or GI tract. How-
ever, neither buboes nor a primary skin ulcer appear.
Penetration is followed by lymphatic spread, facultative
intracellular growth in macrophages, and blood and or-
gan invasion. The symptoms are systemic with fever,
chills, sweats, loss of appetite, backache, headache, and
sometimes lymphadenopathy. The fever usually peaks
in the evening and slowly returns to normal by morning.
The slow rise in temperature during the day, declining
at night, has led to its other name, undulant fever.
These symptoms can last from months to years, but for-
tunately the disease is rarely fatal.
Diagnosis of active disease is best made by culture of
the organism from the blood, bone marrow, liver, or
lymph nodes. Serologic examination that demonstrates
elevated
anti-Brucella
antibodies suggests active dis-
ease. A skin test (with brucellergin) similar to that for
tularemia is available, but a positive result only indi-
cates exposure to the organism and does not prove that
there is active brucellosis.
Pasteurella multocida
This organism is a gram-negative zoonotic organism.
However, it is NOT a facultative intracellular organ-
ism!!!! This bacterium colonizes the mouths of cats
much in the same way that
Streptococcus viridans colo-
nizes the human nasopharynx. It also causes disease in
other mammals and birds.
Fig. 11-5. Cat chasing a bird in a"Pasteur."
76
Figure 11-5
This bacterium causes the most frequent wound in-
fection following a cat or dog bite. When a patient comes
in with a cat or dog bite (or scratch), it is important not
to close the wound with sutures. A closed wound creates
a pleasant environment for
Pasteurella multocida
growth, and the resulting infection can invade local
joints and bones. Treat infected patients with penicillin
or doxycycline.
Fig. 11-6. Summary of zoonotic gram-negative rods.
Recommended Review Articles:
Gill V, Cunha B. Tularemia Pneumonia; Seminars in Respi-
ratory Infections, Vol 12, No. 1; 1997; 61-67.
Titball R, Leary S. Plague. British Medical Bulletin 1998;54:
625-633.
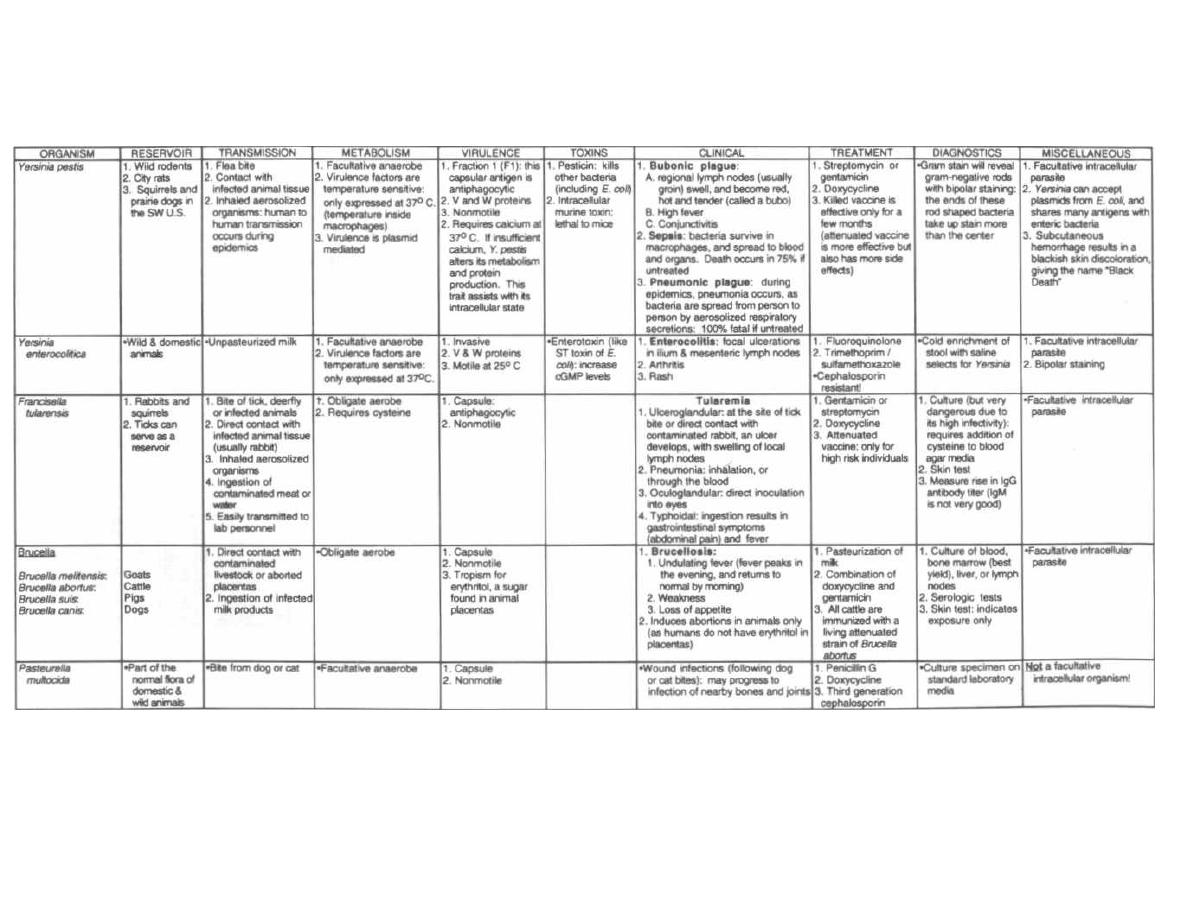
Figure 11-6
ZOONOTIC GRAM-NEGATIVE RODS
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple ©MedMaster

CHAPTER 12.
CHLAMYDIA, RICKETTSIA, AND FRIENDS
Chlamydia and Rickettsia are 2 groups of gram-negative
bacteria that are obligate intracellular parasites. This
means they can survive only by establishing "residence"
inside animal cells. They need their host's ATP as an en-
ergy source for their own cellular activity. They are
energy parasites, using a cell membrane transport
system that steals an ATP from the host cell and spits
out an ADP. Both Chlamydia and Rickettsia have this
ATP/ADP translocator. They differ in that Rickettsia
can oxidize certain molecules and create ATP (via ox-
idative phosphorylation) while Chlamydia does not ap-
pear to have this cytochrome system and in fact has no
mechanism for ATP production. The obligate intracellu-
lar existence brings up 2 questions:
Q: How do we grow and isolate these creatures when
nonliving media do not contain ATP???
A: Indeed, the obligate intracellular existence makes
it impossible to culture these organisms on nonliving ar-
tificial media. However, we can inoculate Chlamydia or
Rickettsia into living cells (most commonly chick em-
bryo yolk sac or cell culture).
Q: Are these bacteria really viruses, since they
are very tiny and use the host's cell for their own
reproduction????
A: Although Chlamydia and Rickettsia share a few
characteristics with viruses (such as their small size
and being obligate intracellular parasites), they have
Figure 12-1 COMPARISON OF CHLAMYDIA
AND RICKETTSIA WITH BACTERIA AND
VIRUSES
78
both RNA and DNA (while viruses have either DNA or
RNA). Also, unlike viruses they both synthesize their
own proteins and are sensitive to antibiotics.
Fig. 12-1.
Comparison of Chlamydia and Rickettsia
with bacteria and viruses.
Chlamydia and Rickettsia cause many distinct hu-
man diseases. Chlamydia spreads by person-to-person
contact, while Rickettsia spreads by an arthropod vector.
CHLAMYDIA
Chlamydia is extremely tiny. It is classified as gram-
negative because it stains red with Gram stain tech-
nique and has an inner and outer membrane. Unlike
other gram-negative bacteria, it does not have a pepti-
doglycan layer and has no muramic acid.
Fig. 12-2.
Chlamydia wearing his CLAM necklace
next to a herpes virus demonstrating that Chlamydia
is about the same size as some of the large viruses.
Chlamydia is especially fond of columnar epithelial
cells that line mucous membranes. This correlates well
with the types of infection that Chlamydia causes, in-
cluding conjunctivitis, cervicitis, and pneumonia.
Bacteria
Chlamydiae
and
rickettsiae
Viruses
Size(nm.)
300-3000
350
15-350
Obligatory
intracellular
parasites
No
YES
YES
Nucleic acids RNA & DNA RNA & DNA RNA Q8 DNA
Reproduction Fission
Complex
cycle with
fission
Synthesis and
assembly
Antibiotic
sensitivity
YES
YES
NO
Ribosomes
YES
YES
NO
Metabolic
enzymes
YES
YES
NO
Energy
production
YES
NO
NO
CHAPTER 12. CHLAMYDIA, RICKETTSIA,
AND FRIENDS
Figure 12-2
The Chlamydia
life cycle is complex as the bacteria
exist in 2 forms:
1) Elementary body (EB): This is a metabolically
inert (does not divide), dense, round, small (300 nm.), in-
fectious particle. The outer membrane has extensive
disulfide bond cross-linkages that confer stability for ex-
tracellular existence.
Fig. 12-3.
Think of the elementary body as an ele-
mentary weapon like the cannon ball, fired from host
cell to host cell, spreading the infection.
2) Initial body (also called reticulate body): Once
inside a host cell the elementary body inhibits phago-
some-lysosome fusion, and grows in size to 1000 nm. Its
RNA content increases, and binary fission occurs, form-
Figure 12-3
79
ing the initial body (IB). Although the IB synthesizes its
own DNA, RNA, and proteins, it requires ATP from the
host. Therefore,
Chlamydia
is considered an energy par-
asite as well as an intracellular parasite.
Fig. 12-4.
The
Chlamydia
life cycle:
A) The infectious particle is the elementary body (EB).
The EB attaches to and enters (via endocytosis) colum-
nar epithelial cells that line mucous membranes.
B) Once within an endosome, the EB inhibits phago-
some-lysosome fusion and is not destroyed. It trans-
forms into an initial body (IB).
C) Once enough IBs have formed, some transform back
i nto EB.
D) The life cycle is completed when the host cell liber-
ates the elementary body (EB), which can now infect
more cells.
There are 3 species of
Chlamydia. Chlamydia tra-
chomatis
primarily infect the eyes, genitals, and lungs;
Chlamydia psittaci
and
Chlamydia pneumonia
only
i nfect the lungs. All are treated with tetracycline or
erythromycin.
Fig. 12-5.
Chlamydial diseases.
Chlamydia trachomatis
Fig. 12-6.
Chlamydia trachomatis
primarily infects
the eyes and genitals. Picture a flower child with
groovy clam eyeglasses and a clam bikini.
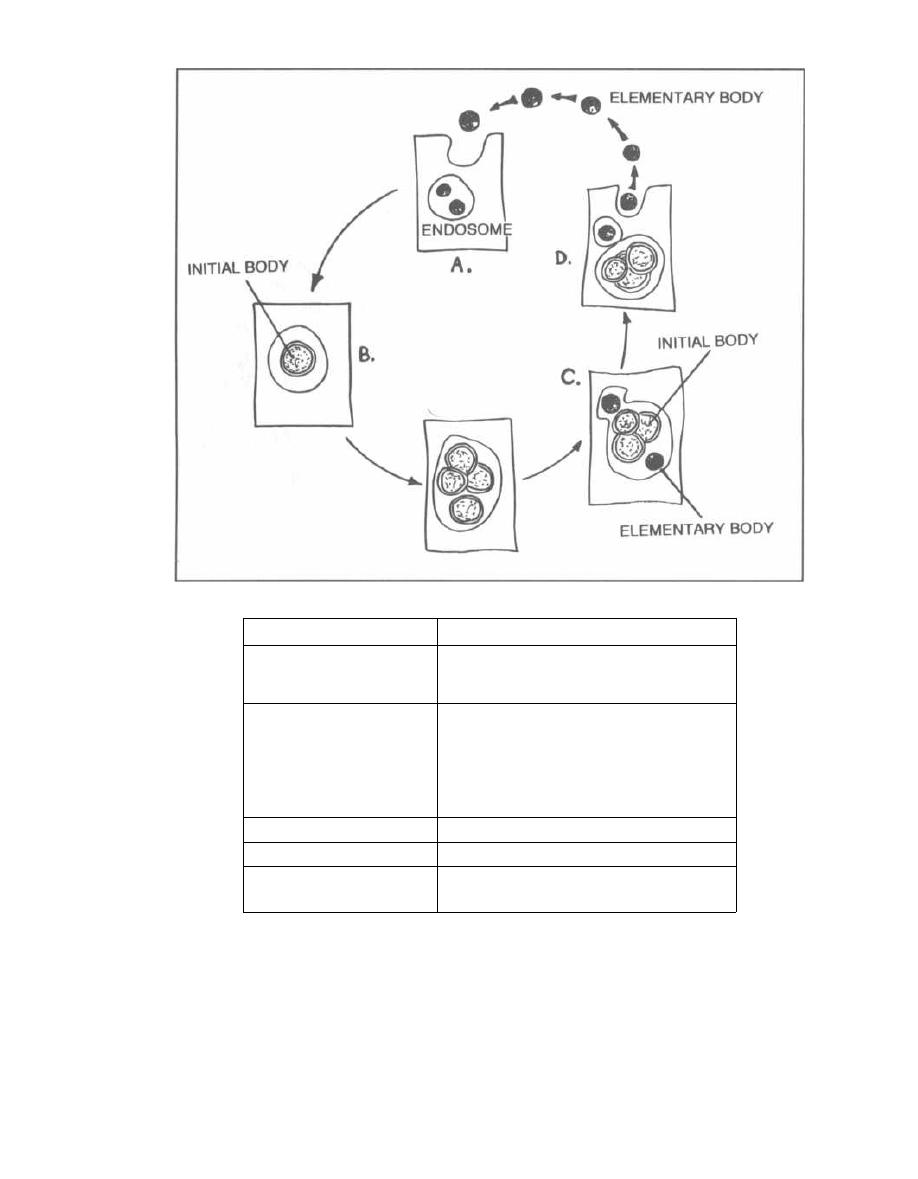
Figure 12-4
CHAPTER 12.
CHLAMYDIA, RICKETTSIA,
AND FRIENDS
Figure 12-5
CHLAMYDIAL DISEASES
Trachoma
hand transfer of infected eye secretions and by sharing
contaminated clothing or towels. Blindness develops
slowly over 10-15 years.
Fig. 12-7.
The conjunctival infection causes inflam-
mation and scarring. Scar traction (trachtion for trach-
oma) pulls and folds the eyelid inward so that the
eyelashes rub against the conjunctiva and cornea,
which causes corneal scarring, secondary bacterial in-
Chlamydia trachomatis is responsible for trachoma,
a type of chronic conjunctivitis that is currently the
leading cause of preventable blindness in the world.
It is a disease of poverty, prevalent in underdeveloped
parts of the world. In the U. S., Native Americans are
the group most frequently infected. Children act as the
main reservoir, and transmission occurs by hand-to-
80
Species
Disease
Chlamydia trachomatis
serotypes A, B, & C
T
rachoma (a leading cause of
blindness in the world)
serotypes D thru K
1. Inclusion conjunctivitis (usually in
newborns, contracted in the birth
canal)
2. Infant pneumonia
3. Cervicitis
4. Nongonococcal urethritis in men
serotypes Lt, L2, L3
L
ymphogranuloma venereum (LGV)
Chlamydia psittaci
A
typical pneumonia
Chlamydia
pneumoniae
Serogroup TWAR
.Atypical pneumonia
Figure 12-6
fections, and ultimately blindness. Simple treatment
with topical tetracycline prevents this illness.
Inclusion Conjunctivitis
As Chlamydia trachomatis
is the most common sexu-
ally transmitted disease in the U. S., it is not surprising
that many babies delivered through birth canals in-
fected with this organism develop inclusion conjunc-
tivitis. Conjunctival inflammation with a purulent
yellow discharge and swelling of the eyelids usually
arises 5-14 days after birth. In the U. S., all newborns
are given erythromycin eye drops prophylactically.
Diagnosis is made by demonstrating basophilic in-
tracytoplasmic inclusion bodies in cells taken from
scrapings of the palpebral conjunctival surface. These
inclusion bodies are collections of initial bodies in the cy-
toplasm of the conjunctival cells.
Infant Pneumonia
A baby's passage through an infected birth canal may
also lead to a chlamydial pneumonia, which usually oc-
curs between 4-11 weeks of life. Initially, the infant de-
velops upper respiratory symptoms followed by rapid
breathing, cough, and respiratory distress.
Diagnosis is made clinically, and the diagnosis can be
later confirmed by the presence of anti-chlamydial IgM
CHAPTER 12.
CHLAMYDIA, RICKETTSIA,
AND FRIENDS
81
Figure 12-7
antibodies and/or demonstration of
Chlamydia tra-
chomatis
in clinical specimens. Treat with oral ery-
thromycin.
Urethritis
Urethritis, an infection of the urethra, is usually con-
tracted sexually.
Neisseria gonorrhoeae
is the most fa-
mous bacterium causing urethritis, but not the most
common. Urethritis that is not caused by
Neisseria gon-
orrhoeae
is called nongonococcal urethritis (NGU), and
is thought to be the most common sexually transmit-
ted disease. NGU is predominantly caused by
Chlamy-
dia trachomatis
and
Ureaplasma urealyticum.
Many patients with NGU are asymptomatic. Symp-
tomatic patients develop painful urination (dysuria)
along with a thin to thick, mucoid discharge from the
urethra. It is impossible clinically to differentiate gono-
coccal urethritis from NGU and they often occur to-
CHAPTER 12.
CHLAMYDIA, RICKETTSIA,
AND FRIENDS
gether as a mixed infection. These mixed infections are
discovered when patients are treated only with a peni-
cillin family antibiotic and don't get better. Penicillins
treat the gonorrhea, but are ineffective against Chlamy-
dia trachomatis. Remember that Chlamydia trachoma-
tis has no peptidoglycan layer, which is the target for
penicillin.
Therefore, all patients diagnosed with urethritis are
empirically treated with antibiotics to cover Neisseria
gonorrhoeae, Chlamydia trachomatis, and Ureaplasma
urealyticum. A commonly used treatment regimen in-
volves a single dose of intramuscular ceftriaxone (a
third-generation cephalosporin that is extremely effec-
tive against Neisseria gonorrhoeae) followed by a 7-day
course of oral doxycycline or 1 oral dose of azithromycin
(which covers both Chlamydia trachomatis and Urea-
plasma urealyticum). (See Chapter 17, page 131.)
While the patient is on empiric antibiotics, diagnostic
tests are performed to determine which organism is re-
sponsible. The diagnosis of chlamydial NGU is a bit
roundabout because the bacteria are too small to visu-
alize with the Gram stain and cannot be cultured on
nonliving media. If the Gram stain reveals polymor-
phonuclear leukocytes but NO intracellular or extracel-
lular gram-negative diplococci (that is, NO Neisseria
gonorrhoeae ), a diagnosis of NGU is likely. If cultures
fail to grow the Neisseria gonorrhoeae, then a diagnosis
of NGU is further supported. The discharge can also be
smeared on a slide and sent for a chlamydial comple-
ment fixation test for absolute confirmation.
Cervicitis and Pelvic Inflammatory Disease
(PID)
The cervix is a frequent site for Chlamydia tra-
chomatis infection. The inflamed cervix appears red,
swollen, and has a yellow mucopurulent endocervical
discharge. This infection can spread upwards to involve
the uterus, fallopian tubes, and ovaries. This infection,
which can be caused by both Chlamydia trachomatis
and Neisseria gonorrhoeae, is called pelvic inflamma-
tory disease (PID).
Women with PID often develop abnormal vaginal dis-
charge or uterine bleeding, pain with sexual intercourse
(dyspareunia), nausea, vomiting, and fever. The most
common symptom is lower abdominal pain. The in-
flamed cervix, uterus, tubes, and ovaries are very
painful. Some medical slang emphasizes this. Women
are observed to have the "PID shuffle" (small, wide-
based steps to minimize shaking of abdomen). With
movement of the cervix on bimanual vaginal examina-
tion the patient may exhibit the "Chandelier sign" (cer-
vical motion tenderness is so severe that the patient
leaps to the chandelier).
PID often results in fallopian tube scarring, which
can cause infertility, tubal (ectopic) pregnancy, and
82
chronic pelvic pain. It is estimated that 1 million women
suffer from PID every year in the U.S. and 25% of them
will become infertile. In one prospective study (We-
strom, 1992), tubal occlusion leading to infertility oc-
curred in 8% of women after 1 episode of PID, 19.5%
after 2 episodes, and 40% after 3 episodes. Likewise, the
risk of ectopic pregnancy and chronic pelvic pain in-
creases with recurrent PID.
Chlamydia trachomatis is particularly dangerous as
it often causes asymptomatic or mild PID that goes un-
diagnosed and untreated, yet can still lead to infertility.
Fig. 12-8.
Infected fallopian tubes scar easily, which
can result in infertility. The silent sinister CLAM
(Chlamydia
trachomatis) causes asymptomatic PID
that can lead to infertility.
A simple shot of ceftriaxone and 14 days of oral
doxycycline will vanquish PID.
(McCormack, 1994)
Epididymitis
Chlamydial epididymitis can develop in men with
urethritis and presents clinically as unilateral scrotal
swelling, tenderness, and pain, associated with fever.
Other Complications of
Chlamydial Infection
Chlamydia trachomatis is also linked to Reiter's
syndrome, an inflammatory arthritis of large joints,
Figure 12-8

that commonly occurs in young men between the ages of
20 and 40. Inflammation of the eyes (uveitis and con-
junctivitis) and urethritis also occur. However, other in-
fectious agents may also precipitate this syndrome.
Fitz-Hugh-Curtis syndrome is an infection of the
liver capsule with symptoms of right upper quadrant
pain that can occur in men and women. This syndrome
is associated with either chlamydial or gonococcal in-
fection.
Lymphogranuloma Venereum
Lymphogranuloma venereum, another sexually
transmitted disease caused
by Chlamydia trachomatis,
(serotypes L1,
L2
and L3) starts with a painless papule
(bump) or ulceration on the genitals that heals sponta-
neously. The bacteria migrate to regional lymph nodes,
which enlarge over the next 2 months. These nodes be-
come increasingly tender and may break open and drain
pus (see Chapter 10, page 69).
Chlamydia psittaci
(Psittacosis)
Chlamydia psittaci
infects more than 130 species of
birds, even pet parrots. Humans are infected by inhaling
Chlamydia-laden
dust from feathers or dried-out feces.
This infection is an occupational hazard for breeders of
carrier pigeons, veterinarians, and workers in pet-shops
or poultry slaughterhouses. Infection results in an atyp-
ical pneumonia called psittacosis, which occurs 1-3
weeks after exposure.
Atypical Pneumonia
Pneumonia caused by viruses,
Mycoplasma pneumo-
niae, Chlamydia psittaci,
and
Chlamydia pneumoniae,
are called atypical pneumonias because the pneu-
monia is very different from a typical bacterial pneu-
monia caused
by Streptococcus pneumonia.
Patients
with atypical pneumonia present with fever, headache,
and a dry hacking cough without production of yellow
sputum. The lung exam is surprisingly normal with
only a few crackles heard with the stethoscope. The
chest X-ray may have patches or streaks of infiltrate.
In contrast, a patient with a streptococcal pneumonia
appears very sick, coughs up gobs of pus, and has a
lobe of the lung socked in with white blood cells and
debris that can be heard on physical exam and seen on
chest X-ray.
Fig. 12-9. A man with atypical pneumonia caused by
Chlamydia psittaci.
He has a bird with a CLAM neck-
lace, PSITTING on his shoulder.
CHAPTER 12.
CHLAMYDIA, RICKETTSIA,
AND FRIENDS
83
Figure 12-9
Chlamydia pneumoniae
(strain TWAR)
Chlamydia pneumonia
TWAR is a recently identified
species of
Chlamydia,
which is transmitted from person
to person by the respiratory route and causes an atypi-
cal pneumonia in young adults worldwide (along with
Mycoplasma pneumoniae).
TWAR is an acronym for its
original isolation in Taiwan and Acute Respiratory.
RICKETTSIA
Rickettsia
is a small, gram-negative, non-motile, rod-
to coccoid-shaped bacterium. It is similar to
Chlamydia
in that they both are the size of large viruses. Both are
obligate intracellular energy parasites (they steal ATP).
However,
Rickettsia
differs from
Chlamydia
in a num-
ber of ways:
1) Rickettsia
requires an arthropod vector (except for
Q fever).
Fig. 12-10. Ricky the riding
Rickettsia
loves to
travel. He rides a tick in Rocky Mountain spotted fever,
a louse in epidemic typhus, and a flea in endemic typhus.
2) Rickettsia
replicates freely in the cytoplasm, in
contrast to
Chlamydia,
which replicates in endosomes
(inclusions).
3)
Rickettsia
has a tropism for endothelial cells that
line blood vessels
(Chlamydia
likes columnar epithe-
lium).
4) They cause different diseases!!! Most
Rickettsia
cause rashes, high fevers, and bad headaches.
Some
Rickettsia
share antigenic characteristics with
certain strains of
Proteus vulgaris
bacteria. It is purely
coincidental that they have the same antigens.
Proteus
CHAPTER 12.
CHLAMYDIA, RICKETTSIA, AND FRIENDS
Figure 12-10
is not involved at all in rickettsial disease. The Proteus
vulgaris strains that share these common antigens are
designated OX-2, OX-19, and OX-K.
The Weil-Felix reaction is a classic test that uses
these cross-reacting Proteus vulgaris antigens to help
confirm a diagnosis of a rickettsial infection. This test
is done by mixing the serum of a patient suspected of
having a rickettsial disease, with antigens from speci-
fic strains of Proteus vulgaris. If the serum has anti-
rickettsial antibodies, latex beads coated with Proteus
antigens will agglutinate, indicating a positive Weil-
Felix test. Comparison of the laboratory results with
Fig. 12-11 can even help distinguish specific rickettsial
diseases. For example, when this test is performed on a
patient with signs and symptoms of a scrub typhus in-
fection, a negative OX-19 and OX-2 along with a posi-
tive OX-K is confirmatory.
8 4
Fig.
12-11.
Antigenic
differences
among the
Rickettsiae.
Diagnosis of a rickettsial infection can also be made
with specific serologic tests documenting a rise in anti-
Rickettsial antibody titers over time. These include the
indirect immunofluorescence test (IFA), the comple-
ment fixation test (CF), and the enzyme-linked immu-
nosorbent assay (ELISA). These tests can specifically
identify species and even subspecies.
Therapy for all rickettsial diseases consists primarily
of doxycycline and chloramphenicol.
Rickettsia rickettsia
( Rocky Mountain Spotted Fever)
Ricky is riding a wood tick

Figure 12-12
CHAPTER 12.
CHLAMYDIA, RICKETTSIA,
AND FRIENDS
Figure 12-11
WEIL-FELIX
Fig. 12-12. Rocky Mountain spotted fever presents
organism,
Rickettsia rickettsia.
This disease is char-
within a week after a person is bitten by either the wood
acterized by fever, conjunctival redness, severe head-
tick
Dermacentor andersoni
or the dog tick
Dermacentor
ache, and a rash that initially appears on the wrists,
v
ariabilis.
Both of these ticks transmit the causative
ankles, soles and palms and later spreads to the trunk.
85
Disease
Weil-Felix
OX-19
Ox-2
OX-K
Rocky Mountain
spotted fever
+
+
-
Rickettsial pox
-
-
-
Epidemic typhus
+
-
-
Endemic typhus
+
-
-
Brill-Zinsser disease
+1-
-
-
Scrub typhus
-
-
+
Trench fever
-
-
-
Q fever
-
-
-
This figure illustrates the spotted Rocky Mountains
behind a boy with headache, fever, palmar rash, and
tick infestation.
Rocky Mountain spotted fever is more common in the
southeastern U. S. tick belt than in the Rocky Mountain
region. This disease should be called Appalachian spot-
ted fever, as most cases currently occur in the south At-
lantic and south central states such as North Carolina,
South Carolina, Tennessee, and Oklahoma. However,
cases have been reported in nearly every state.
The organisms proliferate in the endothelial lining of
small blood vessels and capillaries, causing small hem-
orrhages and thrombi. The inflammation and damage to
small blood vessels explains the conjunctival redness
and skin rash. Although this disease often resolves in
about 3 weeks, it can progress to death (especially when
antibiotic therapy is delayed).
Since the tick transmits this bacteria during its 6-10
hours of feeding, early discovery and removal of ticks
will prevent infection (Spach, 1993).
Rickettsia akari
( Rickettsialpox)
Ricky is riding a mite....
Fig. 12-13.
Rickettsia akari causes rickettsialpox
and is transmitted to humans via mites that live on
house mice. Imagine Ricky, with pox marks, playing
Atari (old type of Nintendo) with his rodent friend
matey mouse.
Rickettsialpox is a mild, self-limited, febrile disease
that starts with an initial localized red skin bump
(papule) at the site of the mite bite. The bump turns into
a blister (vescicle) and days later fever and headache de-
velop, and other vescicles appear over the body (similar
to chickenpox). Although this disease is self-limiting,
CHAPTER 12. CHLAMYDIA, RICKETTSIA, AND FRIENDS
Figure 12-13
8 6
there is a dramatic response to doxycycline. Elimina-
tion of nearby rodents, which can serve as a reservoir for
Rickettsia akari, is i mportant in preventing this disease.
Rickettsia prowazekii
( Epidemic Typhus)
Ricky is riding a louse... .
An epidemic is the sudden onset and rapid spread of
an infection that affects a large proportion of a popula-
tion. Endemic refers to an infectious disease that ex-
ists constantly throughout a population. Two species of
Rickettsia cause typhus. Rickettsia prowazekii causes
an epidemic form, while Rickettsia typhi is responsible
for endemic typhus. Although they have different reser-
voirs and vectors, these are closely related bacteria that
cause a similar disease, and infection with one confers
immunity to the other!!
Fig. 12.14.
Prowazekia is Prower!!! With war, over-
crowding, and poverty, unsanitary conditions prevail
and lice take control, harboring Rickettsia prowazekii.
The lice transmit the bacteria to humans, causing epi-
demic typhus.
This disease wiped out a third of Napoleon's army
when he advanced on Moscow in 1812, and was respon-
sible for more than 3 million Russian deaths in World
War I. The last epidemic in the U. S. occurred more than
70 years ago. Currently, flying squirrels serve as a
reservoir in the southern U. S. Sporadic cases occur
when lice or fleas from infected squirrels bite humans.
Clinically, epidemic typhus is characterized by an
abrupt onset of fever and headache following a 2-week
incubation period. Small pink macules appear around
the fifth day on the upper trunk and quickly cover the
entire body. In contrast to Rocky Mountain spotted
fever, this rash spares the palms, soles, and face. The
patient may become delirious or stuporous. Since Rick-
ettsia invade the endothelial cells of blood vessels, there
is an increased risk of blood vessel clotting leading to
gangrene of the feet or hands. This disease will often re-
solve by 3 weeks, but occasionally is fatal (especially in
older patients).
Diagnosis would be easy during an epidemic. The
poor doctor with Napoleon's retreating forces surely be-
came an expert diagnostician of louse-borne typhus! It
is the sporadic case in the southern U.S., transmitted
from flying squirrels to humans by louse or flea bites,
that is unexpected and thus difficult to diagnose. Close
contact with the flying squirrel vector should raise sus-
picion.
Besides tetracycline and chloramphenicol, improved
sanitation and eradication of human lice will help con-
trol epidemics.

CHAPTER 12. CHLAMYDIA, RICKETTSIA,
AND FRIENDS
Figure 12-14
Brill-Zinsser Disease
For those of you who have referred to the Zinsser Mi-
crobiology textbook, it is interesting to note that Hans
Zinsser is credited with correctly postulating that pa-
tients who recovered without antibiotic therapy from
epidemic louse-borne typhus could still retain the
pathogen Rickettsia prowazekii in a latent state. Occa-
sionally, it breaks out of its latent state to produce Brill-
Zinsser disease. However, symptoms are usually milder
(no skin rash) due to the presence of pre-formed anti-
bodies from the original infection. Diagnosis is made by
demonstrating a rapid early rise in IgG titer specific for
Rickettsia prowazekii, rather than a rapid rise in IgM,
which occurs in the primary infection.
It is always important to completely eradicate Rick-
ettsia prowazekii from your patient with sufficient an-
tibiotic therapy because untreated patients may serve
as a reservoir between epidemics.
Rickettsia typhi
(Endemic or Murine Typhus)
Ricky is riding a flea... .
Endemic flea-borne typhus is similar to epidemic
typhus, yet it is not as severe and does not occur in
epidemics. This disease is caused by Rickettsia typhi.
Rodents serve as the primary reservoir, and the disease
is transmitted to humans via the rat flea, Xenopsylla
8 7
cheopis. ( This flea was also responsible for transmission
of bubonic plague in the past.)
Following a 10-day incubation period, fever, head-
ache, and a flat and sometimes bumpy (maculopapular)
rash develop, just as with epidemic typhus. Although
this disease is milder than that caused by epidemic ty-
phus, it is still very serious.
Treat with doxycycline or chloramphenicol. Control
flea and rat populations. As with the bubonic plague
( Yersinia pestis), we don't want to just kill the rats because
the starving fleas would then all move to bite humans!!!
Rickettsia tsutsugamushi
(Scrub Typhus, or Tsutsugamushi Fever)
Rickettsia tsutsugamushi is found in Asia and the
southwest Pacific. This disease affected soldiers in the
South Pacific during World War II and in Vietnam.
Rickettsia tsutsugamushi is spread by the bite of larvae
(chiggers) of mites. The mites live on rodents, and the
larval chiggers live in the soil.
Fig. 12-15.
Ricky is now a South Pacific sumo
wrestler named Ricky Tsutsugamushi. He is walking in
the scrub (scrub typhus) being bitten by chiggers that
are on his feet and legs.
After a 2-week incubation period, there is high fever,
headache, and a scab at the original bite site. Later

CHAPTER 12. CHLAMYDIA, RICKETTSIA, AND FRIENDS
Figure 12-15
a flat and sometimes bumpy (maculopapular) rash
develops.
Bartonella (formerly Rochalimaea
quintana)
( Trench Fever)
Trench fever is a louse-borne febrile disease that oc-
curred during World War I. The organism responsible
for this disease is Bartonella quintana. Although it is
Rickettsia-like, it has a different genus name because it
is not an obligate intracellular organism.
This disease was spread in the trenches by the body
louse. Infected soldiers developed high fevers, rash,
headache, and severe back and leg pains. After appear-
ing to recover, the soldier would relapse 5 days later.
Multiple relapses can occur but fatalities are rare. The
organism's species name, quintana, reflects the charac-
teristic 5-day interval between febrile episodes.
Notice the similarities here with epidemic typhus
( Rickettsia prowazekii-Prowar Ricky). Both achieve epi-
demic proportions during war, when filth and poor san-
itation lead to lice overgrowth.
FILTH = LICE _
Rickettsia prowazekii (Epidemic typhus) +
Bartonella quintana (trench fever)
Bartonella (formerly Rochalimaea)
henselae
( Cat-scratch Disease and Bacillary Angiomatosis)
Cat-scratch disease occurs following a cat bite or
scratch. A regional lymph node or nodes will enlarge
and the patient may develop low-grade fever and
malaise. The disease usually resolves within a few
months without complications.
A motile, gram-negative rod named Afipia
fells
was originally isolated from affected lymph nodes. How-
8 8
ever, there is now growing evidence that another bac-
terium may be the etiologic agent: Bartonella henselae.
Several studies have now documented high levels of
anti-Bartonella henselae antibodies in patients with
cat-scratch disease. Bartonella henselae may also be re-
sponsible for a disease called bacillary angiomatosis,
which involves a proliferation of small blood vessels in
the skin and organs of AIDS patients (Margileth, 1993;
Zangwill, 1993).
Coxiella burnetii
( Q Fever)
Coxiella burnetii is unique to the Rickettsia because,
like the gram-positive spore formers (Clostridium and
Bacillus), it has an endospore form. This endospore
confers properties to the bacteria that differ from other
Rickettsiae:
1) Resistance to heat and drying: Spores may
contaminate milk products so pasteurization tempera-
tures have to be raised to greater than 60°C to kill the
endospores.
2) Extracellular existence: The spore's resistance
allows extended survival outside a host cell. However,
like Chlamydia and Rickettsia, growth and division
must occur intracellularly using the host's ATP.
3) Non-arthropod transmission: Coxiella burnetii
grows in ticks and cattle. The spores remain viable in
dried tick feces deposited on cattle hides, and in dried
cow placentas following birthing. These spores are
aerosolized and when inhaled cause human disease.
Spore inhalation rather than an arthropod bite causes
Q fever.
4) Pneumonia: Because the spores are inhaled into
the lungs, a mild pneumonia similar to that of a My-
coplasma pneumonia often develops.
Clinically, abrupt onset of fever and soaking sweats
occur 2-3 weeks after infection, along with a pneumo-
nia. This is the only rickettsial disease that causes
pneumonia and in which there is NO rash.
Fig. 12-16.
Visualize Carol Burnett (Coxiella bur-
netti) coughing after inhaling the spores from a cowhide
and dried placental products in the grass.
Ehrlichia canis and chaffeensis
( Ehrlichiosis and Human Ehrlichiosis)
Ehrlichia canis is a disease of dogs. This makes sense
since dogs like to LICK. Dogs get the bacteria from ticks
which jump from dog to dog. The ticks can also bite hu-
mans, transmitting a very close relative of Ehrlichia ca-
nis to humans. This bacterium is now called Ehrlichia
chaffeensis and causes a disease (called human ehrli-

Figure 12-16
CHAPTER 12.
CHLAMYDIA, RICKETTSIA,
AND FRIENDS
89
chiosis) very similar to Rocky Mountain spotted fever.
Patients develop high fever and severe headache, but
rarely (only 20% of the time) rash (Spach, 1993).
Fig.
12-17.
Summary chart
of
Chlamydia
and
Rickettsia.
References
Margileth AM, Hayden GF. Cat Scratch Disease From Feline
Affection to Human Infection. N Engl J Med 1993;
329:53-54.
McCormack WM. Current Concepts: Pelvic Inflammatory Dis-
ease. N Engl J Med 1994;330:115-119.
Spach DH, et al. Tick-borne diseases in the United States.
N Engl J Med 1993;329:936-947.
Westrom L, et. al. Pelvic inflammatory disease and fertility: a
cohort study of 1,844 women with laparoscopically verified
disease and 657 control women with normal laparoscopic
results. Sex Transm Dis 1992;19:185-92.
Zangwill KM, et al. Cat Scratch Disease in Connecticut: Epi-
demiology, Risk Factors, and Evaluation of a New Diag-
nostic Test. N Engl J Med 1993;329:8-13.
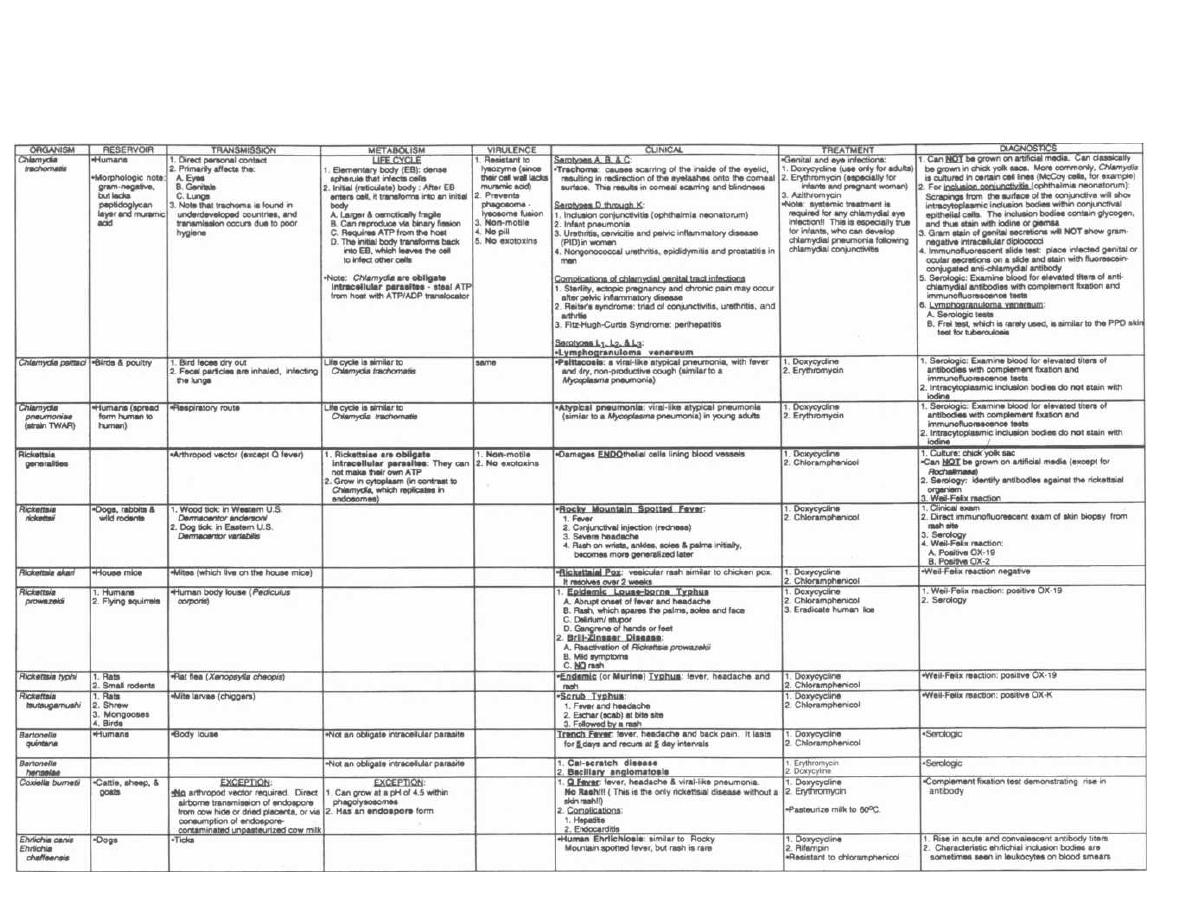
Figure 12-17
GRAM-NEGATIVE OBLIGATE INTRACELLULAR PARASITES: CHLAMYDIA AND RICKETTSIA
Gla
dwin a nd B T
rattle r Clinical
Microbiology Made Ridiculously Sim
ple
Spirochetes are tiny gram-negative organisms that look
like corkscrews. They move in a unique spinning mo-
ion via 6 thin endoflagella called axial filaments,
which lie between the outer membrane and peptidogly-
can layer and wrap around the length of the spirochete.
These organisms replicate by transverse fission.
Spirochetes are a diagnostic problem. They cannot
be cultured in ordinary media, and although they have
gram-negative cell membranes, they are too small to
be seen using the light microscope. Special proce-
dures are required to view these organisms, including
darkfield microscopy, immunofluorescence, and silver
stains. Also, serologic tests help screen for infections
with spirochetes.
Spirochetes are divided into 3 genera: 1) Treponema,
2) Borrelia, and 3) Leptospira.
TREPONEMA
Treponemes produce no known toxins or tissue de-
structive enzymes. Instead, many of the disease mani-
festations are caused by the host's own immune
responses, such as inflammatory cell infiltrates, prolif-
erative vascular changes, and granuloma formation.
Treponema pallidum
( Syphilis)
Treponema pallidum is the infectious agent responsi-
ble for the sexually transmitted disease syphilis. The
number of new cases of syphilis has been increasing
since 1956, with more than 100 thousand cases reported
in 1990 in the U.S. Black heterosexual men and women
living in urban centers are at highest risk for acquiring
syphilis today.
Treponema pallidum enters the body by penetrating
intact mucous membranes or by invading through ep-
ithelial abrasions. Skin contact with an ulcer infected
with Treponema pallidum (even by the doctor's exam-
ining hand) can result in infection. When infection oc-
curs, the spirochetes immediately begin disseminating
throughout the body.
If untreated, patients with syphilis will progress
through 3 clinical stages, with a latent period between
stages 2 and 3.
Fig. 13-1.
Stages of syphilis.
Primary Syphilis
The primary lesion of syphilis is a painless chancre
that erupts at the site of inoculation 3-6 weeks after the
initial contact. Regional nontender lymph node swelling
occurs as well.
CHAPTER 13.
91
SPIROCHETES
Figure 13-1
SYPHILIS: CLINICAL
MANIFESTATIONS
Fig. 13-2.
The chancre can be described as a firm, ul-
cerated painless lesion with a punched-out base and
rolled edges. It is highly infectious, since Treponema
pallidum sheds from it continuously. Think of this skin
ulcer as a small Treponema pallidum resort swimming
pool, with thousands of vacationers swimming within.
The chancre resolves over 4-6 weeks without a scar, of-
ten fooling the infected individual into thinking that the
infection has completely resolved.
Figure 13-2
Secondary Syphilis
Untreated patients enter the bacteremic stage, or sec-
ondary syphilis, often about 6 weeks after the primary
chancre has healed (although sometimes the manifesta-
tions of secondary syphilis occur while the primary chan-
cre is still healing). In secondary syphilis, the bacteria
multiply and spread via the blood throughout the body.
Unlike the single lesion of primary syphilis, the second
stage is systemic, with widespread rash, generalized
lymphadenopathy, and involvement of many organs.
The rash of secondary syphilis consists of small red
macular (flat) lesions symmetrically distributed over
the body, particularly involving the palms, soles, and
mucous membranes of the oral cavity. The skin lesions
can become papular (bumpy) and even pustular.
A second characteristic skin finding of the second
stage is called condyloma latum. This painless, wart-
like lesion often occurs in warm, moist sites like the
Stage
Clinical
Primary stage
P
ainless chancre (ulcer)
Secondary stage
1. Rash on palms and soles
2. Condyloma latum
3. CNS, eyes, bones, kidneys
and/or joints can be involved
Latent syphilis
2
5% may relapse and develop
secondary stage symptoms again
Tertiary stage
1. Gummas of skin and bone
2. Cardiovascular (aortic aneurysm)
3. Neurosyphilis
vulva or scrotum. This lesion, which is packed with spiro-
chetes, ulcerates and is therefore extremely contagious.
Skin infection in areas of hair growth results in
patchy bald spots and loss of eyebrows.
During the secondary stage, almost any organ can be-
come infected (including the CNS, eyes, kidneys, and
bones). Systemic symptoms, such as generalized lym-
phadenopathy, weight loss, and fever, also occur during
this stage.
The rash and condyloma lata resolve over 6 weeks,
and this disease enters the latent phase.
Latent Syphilis
In this stage, the features of secondary syphilis have
resolved, although serologic tests remain positive. Most
patients are asymptomatic during this period, although
about 25% will have one or more relapses and develop
the infectious skin lesions of secondary syphilis. After 4
years, there are generally no more relapses, and this dis-
ease is now considered noninfectious (except in pregnant
women, who can still transmit syphilis to their fetus).
About one-third of untreated patients will slowly
progress from this stage to tertiary syphilis. The rest
will remain asymptomatic.
Tertiary Syphilis
Tertiary syphilis generally develops over 6-40 years,
with slow inflammatory damage to organ tissue, small
blood vessels, and nerve cells. It can be grouped into 3
general categories: 1) gummatous syphilis, 2) cardio-
vascular syphilis, and 3) neurosyphilis.
1) Gummatous syphilis occurs 3-10 years after the
primary infection in 15%n of untreated patients.
Fig. 13-3. Gummas (Gummy bears) are localized
granulomatous lesions which eventually necrose and
become fibrotic. These noninfectious lesions are found
mainly in the skin and bones. Skin gummas are pain-
less solitary lesions with sharp borders, while bone le-
sions are associated with a deep gnawing pain. These
will resolve with antimicrobial therapy.
2) Cardiovascular syphilis occurs at least 10
years after the primary infection in 10% of untreated
patients. Characteristically, an aneurysm forms in the
ascending aorta or aortic arch. This
is
caused by chronic
inflammatory destruction of the small arterioles (vasa
vasorum) supplying the aorta itself, leading to necrosis
of the media layer of the aorta. The wall of the aorta
splits as blood dissects through the weakened media
layer. Aortic valve insufficiency and occlusion of the
coronary arteries may also develop as the dissection
spreads to involve the coronary arteries. Antimicrobial
therapy can NOT reverse these manifestations.
CHAPTER 13. SPIROCHETES
92
Figure 13-3
3) Neurosyphilis occurs in about 8% of untreated
cases. The 5 most common presentations of neurosyphi-
lis are:
a) Asymptomatic neurosyphilis: The patient
is clinically normal, but cerebrospinal fluid tests
positive for syphilis.
b) Subacute meningitis: The patient has fever,
stiff neck, and headache. Cerebrospinal fluid analy-
sis reveals a high lymphocyte count, high protein,
low glucose, and positive syphilis tests.
Note that most bacteria cause an acute meningitis
with a high neutrophil count, high protein, and low
glucose.
Treponema pallidum
and
Mycobacterium
tuberculosis
are two bacteria that cause a subacute
meningitis with a predominance of lymphocytes.
c) Meningovascular syphilis: The spirochetes
attack blood vessels in the brain and meninges (cir-
cle of Willis!), resulting in cerebrovascular occlusion
and infarction of the nerve tissue in the brain,
spinal cord, and meninges, causing a spectrum of
neurologic impairments.
d) Tabes dorsalis: This condition affects the
spinal cord, specifically the posterior column and
dorsal roots.
Fig. 13-4. Syphilitic tabes dorsalis involves damage
to the posterior columns and dorsal roots of the spinal
cord. Posterior column damage disrupts vibratory and
proprioceptive sensations, resulting in ataxia. Dorsal
root and ganglia damage leads to loss of reflexes and
loss of pain and temperature sensation.
e) General paresis (of the insane): This is a pro-
gressive disease of the nerve cells in the brain, lead-
ing to mental deterioration and psychiatric
symptoms.

Figure 13-4
The Argyll-Robertson pupil may be present in
both tabes dorsalis and general paresis. The Argyll-
Robertson pupil, caused by a midbrain lesion, constricts
during accommodation (near vision) but does not react
to light. This is also referred to as the "prostitute's
pupil" because the prostitute accommodates but does
not react, and is frequently infected with syphilis.
Fig. 13-5. Overview of primary, secondary, latent,
and tertiary syphilis. Notice the rule of sixes:
Six-Sexual transmission
6 axial filaments
6 week incubation
6 weeks for the ulcer to heal
6 weeks after the ulcer heals, secondary syphilis
develops
6 weeks for secondary syphilis to resolve
66% of latent stage patients have resolution (no ter-
tiary syphilis)
6 years to develop tertiary syphilis (at the least)
Congenital Syphilis
Congenital syphilis occurs in the fetus of an infected
pregnant woman.
Treponema pallidum
crosses the pla-
cental blood barrier, and the treponemes rapidly dis-
seminate throughout the infected fetus. Fetuses that
acquire the infection have a high mortality rate (still-
birth, spontaneous abortion, and neonatal death), and
almost all of those that survive will develop early or late
congenital syphilis.
Early congenital syphilis occurs within 2 years
and is like severe adult secondary syphilis with wide-
spread rash and condyloma latum. Involvement of the
nasal mucous membranes leads to a runny nose called
the "snuffles." Lymph node, liver, and spleen enlarge-
ment, and bone infection (osteitis-seen on X-ray), are
also common afflictions of early congenital syphilis.
Late congenital syphilis is similar to adult tertiary
syphilis except that cardiovascular involvement rarely
occurs:
1) Neurosyphilis is the same as adults and eighth
nerve deafness is common.
CHAPTER 13. SPIROCHETES
93
2) Bone and teeth are frequently involved. Periosteal
(outer layer of bone) inflammation destroys the carti-
lage of the palate and nasal septum, giving the nose a
sunken appearance called saddle nose. A similar in-
flammation of the tibia leads to bowing called saber
shins. The upper central incisors are widely spaced
with a central notch in each tooth ( Hutchinson's
teeth) and the molars have too many cusps (mulberry
molars).
3) Eye disease such as corneal inflammation can
occur.
Interestingly,
Treponema pallidum
infection does not
damage the fetus until the fourth month of gestation, so
treating the mother with antibiotic therapy prior to this
time can prevent congenital syphilis.
Diagnostic Tests
for
Syphilis
Absolute diagnosis during the first and second stages
can be made by direct examination, under darkfield mi-
croscopy, of a specimen from the primary chancre, the
maculopapular rash, or the condyloma latum. Darkfield
microscopy reveals tiny helically-shaped organisms
moving in a corkscrew-like fashion.
Since direct visualization of spirochetes is effective
only during the active stages of primary and secondary
syphilis, serologic tests were developed as a screening
tool. There are 2 types of serologic screening test: non-
specific and specific.
1) Nonspecific treponemal tests: Infection with
syphilis results in cellular damage and the release into
the serum of a number of lipids, including cardiolipin
and lecithin. The body produces antibodies against
these antigens. We therefore quantitatively measure
the titer of the antibodies that bind to these lipids. If a
patient's serum has these antibodies, we suspect that
he/she has syphilis. Since invasion of the cerebrospinal
fluid (CSF) by syphilis also stimulates an increase of
these anti-lipoidal antibodies, we can also perform this
test on the CSF to diagnose neurosyphilis. The two most
common tests employing this technique are the Vene-
real Disease Research Laboratory (VDRL) and
Rapid Plasma Reagin (RPR) test.
So why are these tests nonspecific? It is important to
realize that 1% of adults without syphilis will also have
these antibodies, resulting in a false positive test. For
example, false positive tests often occur in patients who
are pregnant, have an acute febrile illness such as in-
fectious mononucleosis or viral hepatitis, use intra-
venous drugs, or following immunization. Therefore, a
positive nonspecific test must be confirmed with a spe-
cific treponemal antibody test.
2) Specific treponemal tests: While the nonspe-
cific tests look for anti-lipoidal antibodies, the specific
treponemal tests look for antibodies against the spiro-
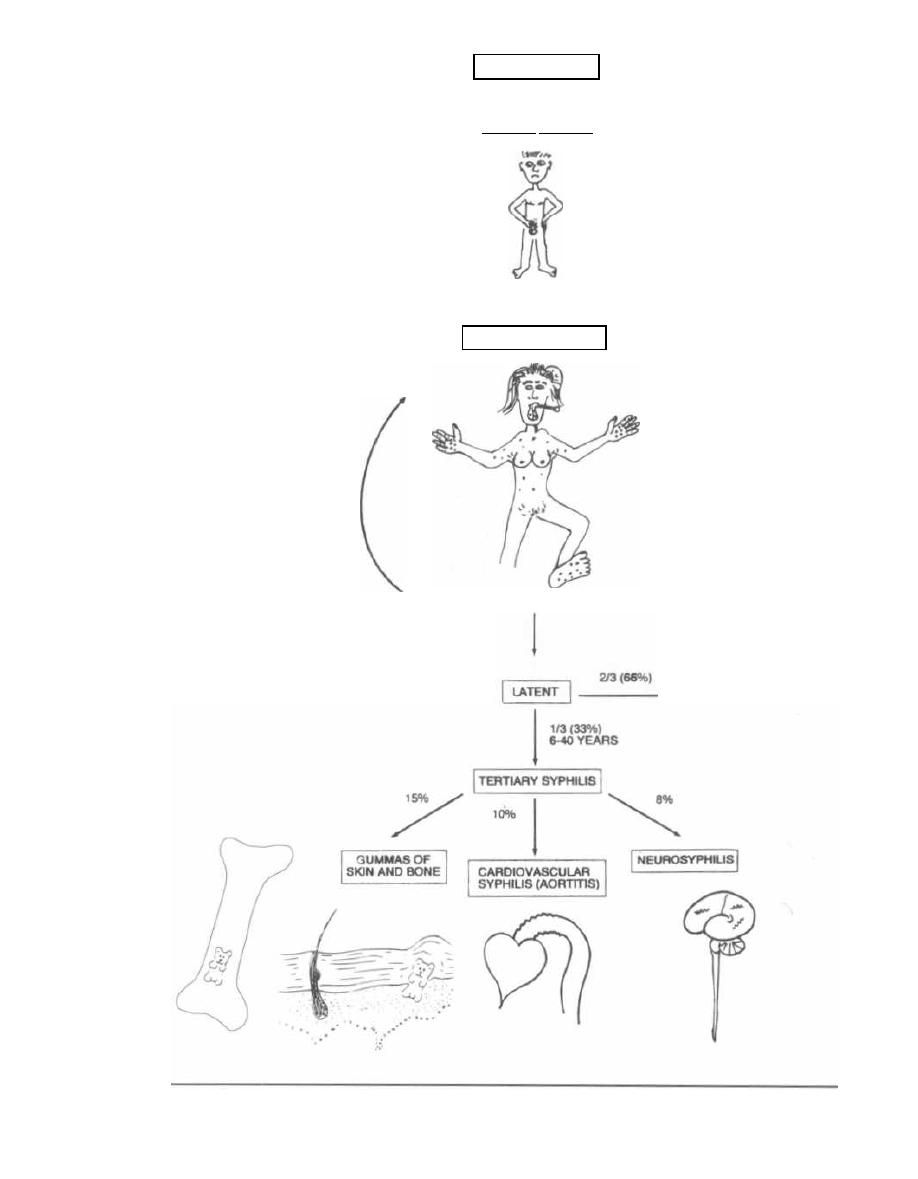
25%
RELAPSE
TO
SECONDARY
SEXUAL CONTACT
PRIMARY SYPHILIS
34 WEEK INCUBATION
6 WEEKS
SECONDARY SYPHILIS
ULCER HEALS AFTER
4-6 WEEKS
SYMPTOMS LAST 2-6 WEEKS
H
AIR LOSS
F
EVER, WEIGHT LOSS
G
ENERALIZED LYMPHADENOPATHY
AND RED RASH (ESPECIALLY PALMS
AND SOLES)
C
ONDYLOMA LATUM
RESOLUTION
Figure 13-5

chete itself. The
Indirect Immunofluorescent Tre-
ponemal Antibody-Absorption (FTA-ABS) test is
the most commonly used specific treponemal test. This
test is performed by first mixing the patient's serum
with a standardized nonpathogenic strain of
Tre-
ponema,
which removes (absorbs) antibodies shared by
both
Treponema pallidum
and the nonpathogenic tre-
ponemal strains (as nonpathogenic strains of
Tre-
ponema
are part of the normal human flora). The
remaining serum is then added to a slide covered with
killed
Treponema pallidum
(as the antigen). Antibodies
that are specific to this organism will subsequently
bind, giving a positive result.
Since we all have antibodies to nonpathogenic strains
of treponemes, the absorption part of the FTA-ABS test
is necessary to cut down on the number of false posi-
tives. Only people who have antibodies specific for the
pathogenic strain of treponemes will elicit a positive re-
action. However, false positives can occur with other
spirochetal infections, such as yaws, pinta, leptospiro-
sis, and Lyme disease.
Treatment
Treponema pallidum is
extremely fragile, and can be
killed easily with heat, drying, or soap and water. Since
syphilis was found to be treatable by raising one's body
temperature, patients in the early 1900's were placed in
a "fever" box (a closed box in the hot sun, with only the
patient's head protruding). Fortunately, the discovery
of
penicillin provided a less hazardous therapy.
The current drug of choice for syphilis is penicillin (the
particular type and dosage of penicillin depends on the
stage of the infection). Penicillin can even cross the pla-
centa and cure congenital syphilis. Patients allergic to
penicillin can be effectively treated with erythromycin
and doxycycline (but doxycycline cannot be used to treat
congenital syphilis, as it is toxic to the fetus).
It is important to realize that reinfection can occur.
This suggests that antitreponemal antibodies are not
protective. Cell-mediated immunity may play a role in
the course of syphilis by inducing the regression of the
lesions of primary and secondary syphilis.
With adequate treatment, the levels of anticardi-
olipin antibodies will decrease, while the levels of spe-
cific antitreponemal antibodies will remain unchanged.
Therefore, a person who is adequately treated will even-
tually manifest (over months to years) a drop in the
VDRL or RPR to nonpositive, while the FTA-ABS will
remain positive.
Fig. 13-6.
Interpretation of syphilis serology.
Jarisch-Herxheimer Phenomenon
Most patients with syphilis will develop an acute
worsening of their symptoms immediately after antibi-
CHAPTER 13. SPIROCHETES
95
Note: VDRL and RPR are very similar tests.
Figure 13-6
SYPHILIS SEROLOGY INTER-
PRETATION
otics are started. Symptoms include a mild fever, chills,
malaise, headache, and muscle aches. The killed organ-
isms release a pyrogen (fever-producing enzyme) that is
thought to cause these symptoms. This self-limiting re-
action, called the Jarisch-Herxheimer phenome-
non, may occur with most spirochetes.
Treponema pallidum
Subspecies
There are 3 subspecies of
Treponema pallidum
(en-
demicum, pertenue, and carateum) that cause non-
venereal disease (endemic syphilis, yaws, and pinta,
respectively). All 3 subspecies cause skin ulcers and
gummas of the skin and bones in children, with the ex-
ception of
Treponema carateum,
which only causes skin
discoloration (no gummas).
Interestingly, these subspecies are morphologically
and genetically identical to
Treponema pallidum,
yet do
not cause the sexually transmitted disease syphilis.
However, the diseases do share many characteristics
with syphilis. Like syphilis, the general pattern of these
diseases involves a primary skin papule or ulcer devel-
oping at the site of inoculation (usually not the genitals).
This is followed by a secondary stage of widespread skin
lesions. The tertiary stage is manifested years later by
gummas of the skin and bones. Unlike tertiary syphilis,
the tertiary stages of the nonvenereal treponemes do
not involve the heart or central nervous system.
The antibodies produced by these infections will give
a positive VDRL and FTA-ABS. One intramuscular in-
jection of long-acting penicillin is curative.
Treponema pallidum
Subspecies
endemicum
( Endemic Syphilis: Bejel)
Endemic syphilis occurs in the desert zones of Africa
and the Middle East and is spread by sharing drinking
VDRL or RPR FTA-ABS
Interpretation
+
+
I
ndicates an active
treponemal infection
+
-
P
robably a false positive
-
+
•
Successfully treated
syphilis
-
-
S
yphilis unlikely, although:
1. Patients with a syphilis
infection who also have AIDS
may be sero-negative
2. Patient recently infected
with syphilis may not have
developed an immune
response yet

and eating utensils. Skin lesions usually occur in the
oral mucosa and are similar to condyloma lata of sec-
ondary syphilis. Gummas of the skin and bone may de=
velop later.
Treponema pallidum
Subspecies
pertenue
(Yaws)
Yaws, a disease of the moist tropics, spreads from per-
son to person by contact with open ulcers. At the initial
site of inoculation a papule appears that grows over
months, becoming wartlike and is called the "mother
yaw." Secondary lesions appear on exposed parts of the
body and years later tertiary gummas develop in the
skin and long bones.
Fig. 13-7.
The tertiary lesions in yaws often cause sig-
nificant disfigurement of the face. Imagine JAWS
( Yaws) taking a bite out of a person's face.
Treponema pallidum
Subspecies
carateum
(Pinta)
Fig. 13-8.
Hispanic person with colored red and blue
skin lesions, saying, "Por favor, no pinta mi cabeza."
Pinta is purely a skin disease limited to rural Latin
America. After infection by direct contact, a papule de-
velops which slowly expands. This is followed by a sec-
ondary eruption of numerous red lesions that turn blue
in the sun. Within a year the lesions become depig-
mented, turning white. These colored lesions look like
someone PAINTED them on.
BORRELIA
The corkscrew-shaped Borrelia are larger than the
Treponema, and therefore can be viewed under a light
microscope with Giemsa or Wright stains.
Figure 13-7
CHAPTER 13. SPIROCHETES
9 6
Figure 13-8
Borrelia cause Lyme disease (Borrelia burgdorferi)
and relapsing fever (caused by 18 other species of Bor
relia). Both of these diseases are transmitted by insect
vectors.
Borrelia burgdorferi
(Lyme Disease)
Lyme disease is seen in the Northeast, Midwest and
northwestern U. S. This is the most commonly reported
tick-borne illness in the U. S.
When walking in the woods during the summer
months, you must be careful of the Ixodes tick. This tiny
creature's bite can transfer the agent for Lyme disease,
Borrelia burgdorferi. It takes greater than 24 hours of
attachment for transfer of the organism, so regular "tick
checks" may help prevent infection.
The animal reservoir for Borrelia burgdorferi in-
cludes the white-footed mouse (as well as other small ro
•
dents) and the white-tailed deer. The Ixodes ticks pick
up the spirochete from these reservoirs and can subse
-
quently transmit them to humans.
CHAPTER 13. SPIROCHETES
Figure 13-9
LYME DISEASE: CLINICAL MANI-
FESTATIONS
Lyme disease has many features that resemble
syphilis, although Lyme disease is NOT sexually trans-
mitted. Both of these diseases are caused by spiro-
chetes. The primary stage in both involves a single,
painless skin lesion (syphilitic chancre and Lyme's ery-
thema chronicum migrans) that develops at the initial
site of inoculation. In both diseases the spirochetes then
spread throughout the body, invading many organ sys-
tems, especially the skin. Both also cause chronic prob-
lems years later (tertiary syphilis and late stage Lyme
disease).
Fig. 13-9.
Like syphilis, Lyme disease has been di-
vided into three stages: 1) early localized stage, 2) early
disseminated stage, and 3) late stage.
Early Localized Stage
The first stage begins about 10 days after the tick
bite and lasts about 4 weeks. It consists of just a skin
lesion at the site of the tick bite (called erythema
chronicum migrans) along with a flulike illness, and
regional lymphadenopathy.
Fig. 13-10.
Erythema chronicum migrans (ECM)
starts off as a red (erythematous) flat round rash,
which spreads out (or migrates) over time (chron-
icum). The outer border remains bright red, while the
center will clear, turn blue, or even necrose. Visualize a
drop of Lyme juice (drawn as drops of spirochetes) land-
ing on the skin and the Lyme acid burning the skin red.
With time, the juice spreads out and the erythematous
lesion spreads, eventually getting so large that there is
not enough juice for the center, so that the center now
has normal-looking skin.
Early Disseminated Stage
Fig. 13-11.
The early disseminated stage involves the
dissemination of
Borrelia burgdorferi
spirochetes to 4
9 7
organ systems: the skin, nervous system, heart, and
joints. Notice that the Lyme juice (drawn as drops of
spirochetes) has begun dripping onto the skin, nervous
system, heart, and joints. This stage can occur after or
at the same time as the first stage.
The skin lesions in this stage are just ECM again,
but this time there are multiple lesions on the body,
and they are smaller (there's just not enough Lyme
juice to make them as large as the one in the primary
stage).
Borrelia burgdorferi
can invade the brain, cranial
nerves, and even motor/sensory nerves. Examples in-
clude meningitis, cranial nerve palsies (especially of
the seventh nerve-a Bell's palsy), and peripheral
neuropathies.
Figure 13-10
Stage
Clinical
Early localized stage
(Stage 1)
E
rythema chronicum migrans (ECM)
Early disseminated stage
(stage 2)
1. Multiple smaller ECM
2. Neurologic: aseptic meningitis,
cranial nerve palsies (Bell's palsy),
and peripheral neuropathy
3. Cardiac: transient heart block or
myocarditis
4. Brief attacks of arthritis of large
joints (knee)
Late stage
(stage 3)
1. Chronic arthritis
2. Encephalopathy
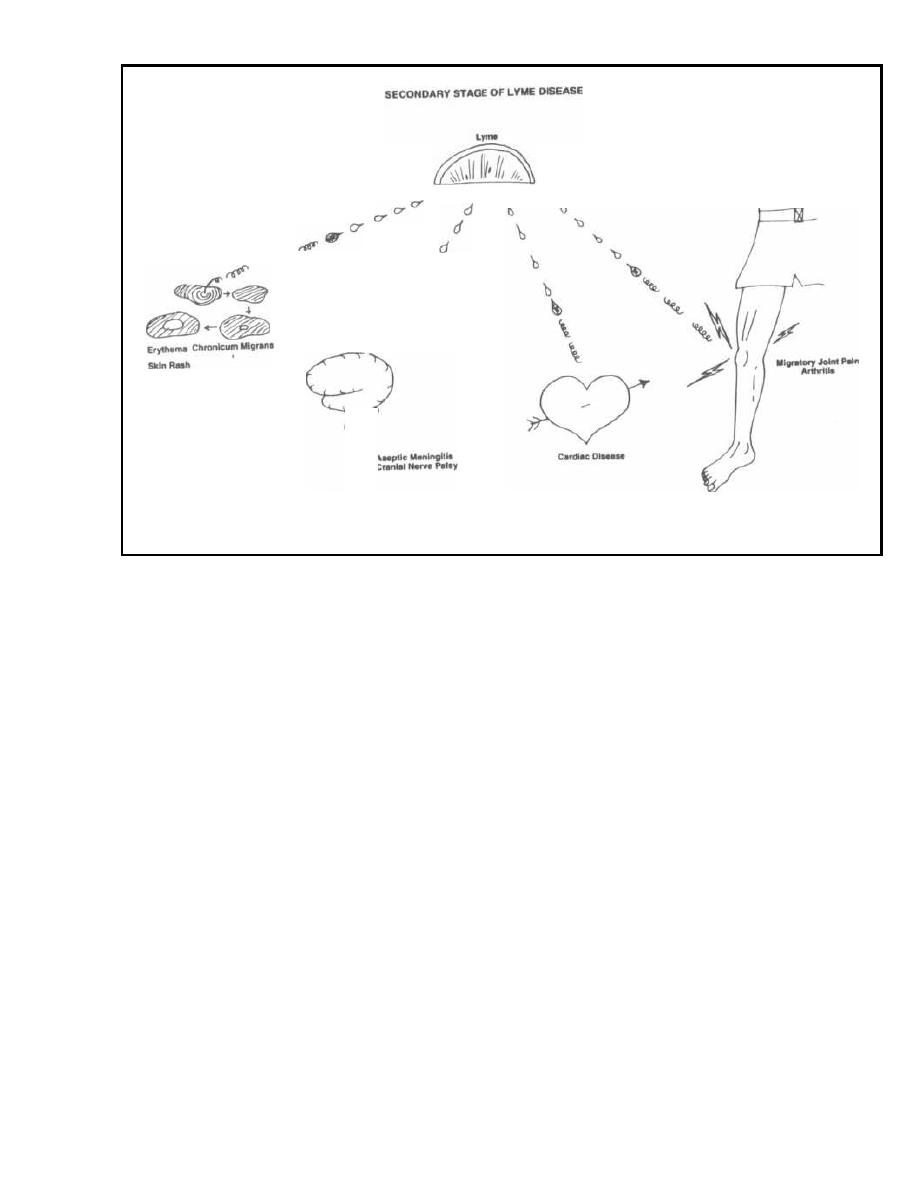
Figure 13-11
Transient cardiac abnormalities occur in about
10%
of
patients. The most common abnormality is atrioventric-
ular nodal block (heart block), and less commonly my-
ocarditis and left ventricular dysfunction. Since the
cardiac lesions usually resolve in a matter of weeks (es-
pecially with antibiotic therapy), a permanent pace-
maker is often unnecessary.
Migratory joint and muscle pain can also occur. About
6 months after infection attacks of arthritis can occur. Large
joints such as the knee become hot, swollen, and painful.
Late Stage
About 10% of untreated patients will develop chronic
arthritis that lasts for more than a year. This usually in-
volves 1 or 2 of the large peripheral joints, such as the
knee. Interestingly, many of these patients have the B-
cell allo-antigen HLA-DR (1 + 4).
Like tertiary syphilis, Lyme disease can lead to
chronic neurologic damage. An encephalopathy can de-
velop characterized by memory impairment, irritability,
and somnolence.
Diagnosis and Treatment
Diagnosis primarily depends on the doctor's recogniz-
i ng the characteristic clinical findings described above
CHAPTER 13. SPIROCHETES
98
in a person who has been exposed to ticks in an area en-
demic for Lyme disease.
If the patient presents with ECM, the leading edge of
the rash can be biopsied and cultured for
Borrelia
burgdorferi.
As culturing this organism from blood and CSF is
very difficult, determination of the levels of
anti-Borre-
lia burgdorferi
antibodies is often helpful in making a
diagnosis. The two most effective techniques are en-
zyme-linked immunosorbent assays (ELISA) and West-
ern immunoblotting.
Doxycycline or penicillin family antibiotics are
currently the most effective antibiotics for treating this
disease. (Spach, 1993)
Two vaccines have recently been developed for Lyme
disease. Both act by passing antibodies through the
individual's blood into the biting tick. The antibodies
neutralize bacteria in the tick before they can be trans-
mitted to the human. The vaccines are ImuLyme and
LYMErix.
Borrelia recurrentis
( Relapsing Fever)
Of 18 different species of
Borrelia
that can cause re-
lapsing fever, only
Borrelia recurrentis
is transmitted to

humans via the body louse
( Pediculus humanus).
The
other
Borrelia
species are transmitted by the tick
Or-
nithodoros.
This tick likes to feed on sleeping campers in
the western U.S., especially those who sleep in rodent-
infested, rustic mountain cabins.
After the
Borrelia
has been transmitted, via the louse
or tick, this bacteria disseminates via the blood. A high
fever develops, with chills, headaches and muscle aches.
Rash and meningeal involvement may follow. With
drenching sweats, the fever and symptoms resolve after
3-6 days. The patient remains afebrile for about S days,
but then relapses, developing similar features for an-
other 3-6 days. Relapses will continue to occur, al-
though they will become progressively shorter and
milder as the afebrile intervals lengthen.
Antigenic Variation: the Key to
Relapsing Fever
Fig. 13-12.
"Why the relapses?" you ask. Well, check
out our friend, Boris the
Borrelia,
who is a master at
the art of "antigenic variation." He is initially well cam-
ouflaged in blood, but antibodies are soon manufac-
tured by the host's immune system. These antibodies
can bind specifically to the
Borrelia
surface proteins
and thereby remove the
Borrelia
from the blood. But
sneaky Boris rapidly changes his surface proteins, so
that the antibodies no longer recognize them. Boris can
now safely proliferate without antibody interference,
resulting in fever. As soon as the immune system rec-
ognizes that there are new foreign proteins in the
blood, it churns out a new set of antibodies that are
specific for Boris's new surface proteins. But Boris is
ready, and quickly changes his surface proteins again.
This antigenic variation allows Boris to continue caus-
ing relapses for many weeks.
Diagnosis is made by drawing blood cultures (culture
on special media) during the febrile periods only (as
blood cultures are often negative when the patient is
afebrile). A Wright's or Giemsa-stained smear of pe-
ripheral blood during febrile periods may reveal the
spirochete between red blood cells. Dark-field mi-
croscopy is also useful.
Doxycycline or erythromycin is the treatment of
choice.
LEPTOSPIRA
Leptospira
are long, thin aerobic spirochetes that are
wound up in a tight coil. They have a hook on one or both
ends, giving them an "ice tongs" appearance. Currently
Leptospira
are divided into 2 species. One of them,
Lep-
tospira interrogans,
causes human disease and has been
divided by serologic tests into 23 serogroups (sub-
groups) and over 240 serovars (sub-subgroups).
CHAPTER 13. SPIROCHETES
99
Figure 13-12
Leptospira
are found all over the world in the urine of
dogs, rats, livestock, and wild animals. These spiro-
chetes can penetrate abraded skin or mucous mem-
branes when humans come in contact with the urine
either directly or by swimming in contaminated water
(usually swallowed).
Clinically, there are 2 phases. In the first or lepto-
spiremic phase the bacteria invade the blood and CSF,
causing an abrupt onset of high spiking temperatures,
headache, malaise, and severe muscle aches (thighs and
lower back). Classically, the conjunctiva are red and the
patient experiences photophobia. After about 1 week,
there is a short afebrile period and then the fever and
earlier symptoms recur. This second or immune
phase correlates with the appearance of IgM antibod-

ies. During the second phase patients may develop
meningismus, and the cerebrospinal fluid (CSF) exam
reveals an elevated white cell count in most patients.
Leptospira interrogans (classically serogroup ictero-
haemorrhagiae, but can be other serogroups) can cause
a more severe illness called Weil's disease, or infec-
tious jaundice, which involves renal failure, hepatitis
with jaundice, mental status changes, and hemorrhage
in many organs.
Diagnosis is made by culturing (on special media)
blood and CSF during the first febrile phase. During the
second phase and months later the organisms can be
cultured from the urine.
The only problem is that treatment should be initi-
ated quickly, before any of the above diagnostic test re-
CHAPTER 13. SPIROCHETES
10 0
suits are available. To arrive at your diagnosis, you
must integrate the clinical history (animal contact or
swimming in areas shared by animals), symptoms sug-
gestive of leptospirosis, and lab tests reflecting the af-
fected organs (elevated liver function tests and protein
in the urine). Treat patients immediately with either
penicillin or doxycycline.
Fig. 13-13.
Summary of the spirochetes.
References
Spach DH, et. al. Tick-borne diseases in the United States. N
Engl J Med 1993;329:936-947.
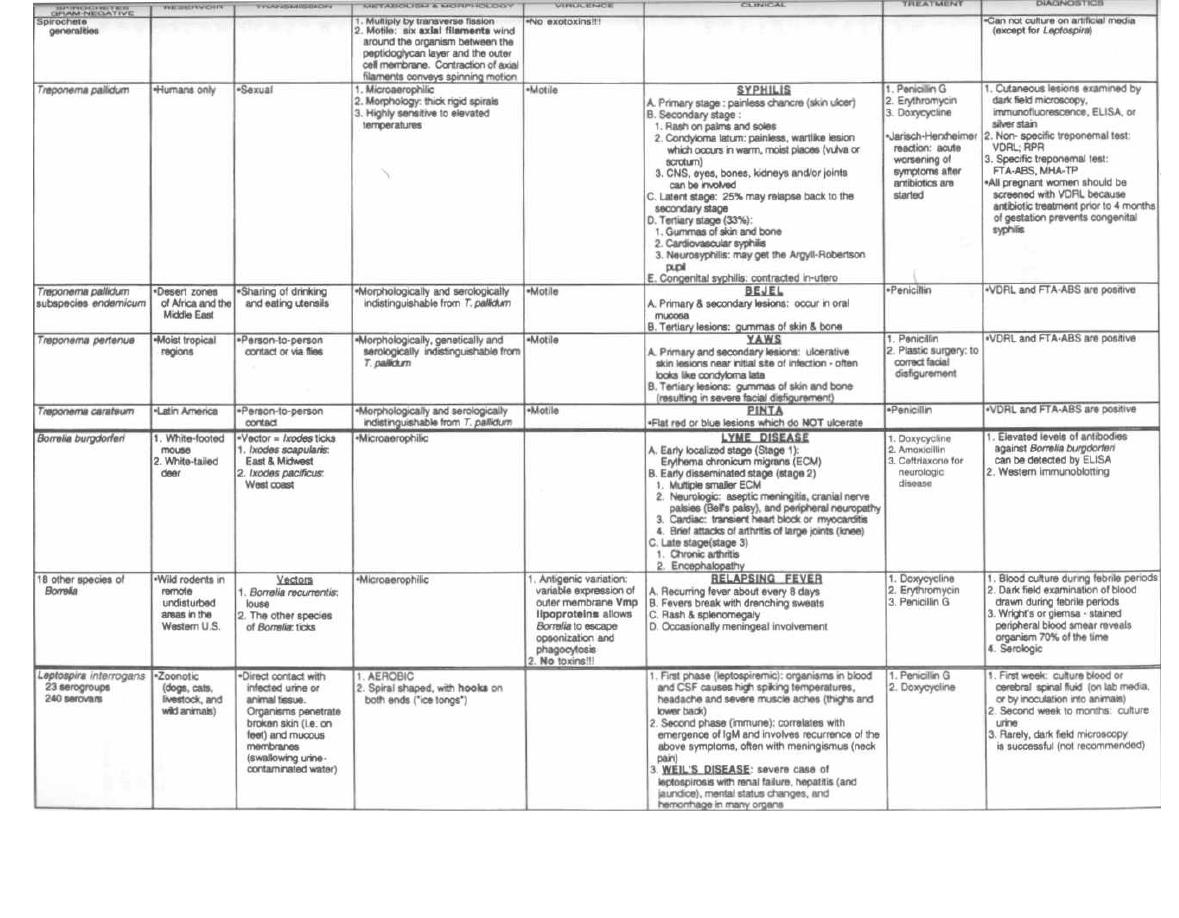
The
Mycobacteria
include 2 species that almost every
one has heard of:
Mycobacterium tuberculosis,
which
causes tuberculosis, and
Mycobacterium leprae,
which
causes leprosy. Humans are the only species infected
with these critters. These organisms are thin rods with
lipid-laden cell walls. This high lipid content makes
them acid-fast on staining. Only
Mycobacteria
and
No-
cardia
are acid-fast.
In the acid-fast stain, a smear of sputum, for exam-
ple, is covered with the red stain carbolfuchsin and
heated to aid dye penetration. Acid alcohol (95% ethanol
and 3% HCl) is poured over the smear, and then a
counter-stain of methylene blue is applied. The cell wall
lipids of the
Mycobacterium
do not dissolve when the
acid alcohol is applied, and thus the red stain does not
wash off. So acid-fast organisms resist decolorization
with acid alcohol, holding fast to their red stain, while
bacteria that are not acid-fast lose the red stain and
take on the blue.
Fig. 14-1. Visualize a fast red sports car to remem-
ber that acid-fast organisms stain red.
Figure 14-1
Mycobacterium tuberculosis
(Tuberculosis)
Worldwide, there are an estimated 10 million new
cases of tuberculosis and 3 million deaths from tuber-
culosis annually. Tuberculosis is currently on the rise in
the U.S., particularly involving the elderly (especially in
nursing homes), AIDS patients, and the urban poor.
ACID-FAST BACTERIA
CHAPTER 14.
MYCOBACTERIUM
102
Persons infected with HIV lack the powerful cell-
mediated immunity necessary to combat tuberculosis.
With the rise in HN infected persons, we are witness-
ing a rise in tuberculosis. About
1
I3
of HIV infected
persons worldwide also harbor
Mycobacterium tubercu-
losis! You will confront this villain again and again in
your future career.
This acid-fast bacillus (rod) is an obligate aerobe,
which makes sense as it most commonly infects the
lungs, where oxygen is abundant.
Mycobacterium tuber-
culosis
grows very slowly, taking up to 6 weeks for visi-
ble growth. The colonies that form lump together due to
their hydrophobic lipid nature, resulting in clumped
colonies on agar and floating blobs on liquid media.
Figure 14-2
There is one class of lipid that only acid-fast or-
ganisms have and that is involved in mycobacterial
virulence-mycosides. The terminology is as follows:
1) Mycolic acid is a large fatty acid.
Fig. 14-2. The chemical structure of mycolic acid,
which is a large fatty acid.
2) Mycoside is a mycolic acid bound to a carbohy-
drate, forming a glycolipid.
3) Cord factor is a mycoside formed by the union of
2 mycolic acids with a disaccharide (trehalose). This my-
coside is only found in virulent strains of
Mycobac-
terium tuberculosis.
Its presence results in parallel
growth of the bacteria, so they appear as cords. Exactly
how the virulence occurs is still unknown, but experi-
ments show that cord factor inhibits neutrophil migra-
tion and damages mitochondria. Its injection into mice
results in the release of tumor necrosis factor (TNF or
Figure 14-3
cachectin), resulting in rapid weight loss. Tuberculosis
in humans is usually a chronic disease with weight
loss that can be mistaken for the cachexia of malig-
nancy. Cord factor might contribute to this weight loss
phenomenon.
4) Sulfatides are mycosides that resemble cord fac-
tor with sulfates attached to the disaccharide: They in-
hibit the phagosome from fusing with the lysosome that
contains bacteriocidal enzymes. The facultative intra-
cellular nature of
Mycobacterium tuberculosis
during
early infection may be partly attributable to the sul-
fatides (see Fig. 2-7).
5) Wax D is a complicated mycoside that acts as an
adjuvant (enhances antibody formation to an antigen)
and may be the part of
Mycobacterium tuberculosis
that
activates the protective cellular immune system.
Fig. 14-3.
To remember the names of the mycosides and
their relationship to
Mycobacterium tuberculosis,
picture
the surfing dude Mike (mycosides). He is WAXING (wax
D) his SUrfboard (sulfatides) and has his surfboard
CHAPTER 14.
MYCOBACTERIUM
10 3
CORD (cord factor) attached to his leg (so as not to lose
his stick). Notice Mike has a cough and some weight loss.
Pathogenesis of Tuberculosis
Mycobacterium tuberculosis
primarily affects the
lung but can also cause disease in almost any other tis-
sue. The way it spreads and damages the body depends
on the host's immune response. The organism and the
immune system interact as follows:
1) Facultative intracellular growth: With the
first exposure (usually by inhalation into the lungs),
the host has no specific immunity. The inhaled bac-
teria cause a local infiltration of neutrophils and
macrophages. Due to the various virulence factors, the
phagocytosed bacteria are not destroyed. They multiply
and survive in the macrophages. The bacteria cruise
through the lymphatics and blood to set up camp in dis-
tant sites. This period of facultative intracellular exis-

tence is usually short-lived because the host rapidly ac-
quires its prime defense against the acid-fast buggers:
cell-mediated immunity.
2) Cell-mediated immunity:
Some of the
macrophages succeed in phagocytosing and breaking
up the invading bacteria. These macrophages then run
toward a local lymph node and present parts of the bac-
teria to T-helper cells. The sensitized T-cells then
multiply and enter the circulation in search of My-
cobacterium tuberculosis. When the T-cells encounter
their antigenic target, they release lymphokines that
serve to attract macrophages and activate them when
they arrive. These activated macrophages can now de-
stroy the bacteria. It is during this stage that the
macrophage attack actually results in local destruction
and necrosis of the lung tissue. The necrosed tissue
looks like a granular creamy cheese and is called
caseous necrosis. This soft caseous center is sur-
rounded by macrophages, multinucleated giant cells,
fibroblasts, and collagen deposits, and it frequently cal-
cifies. Within this granuloma the bacteria are kept at
bay but remain viable. At some point in the future, per-
haps due to a depression in the host's resistance, the
bacteria may grow again.
PPD Skin Test
Following induction of cell-mediated immunity
against Mycobacterium tuberculosis, any additional ex-
posure to this organism will result in a localized de-
layed-type
hypersensitivity
reaction
(type
IV
hypersensitivity). Intradermal injection of antigenic
protein particles from killed Mycobacterium tuberculo-
sis, called PPD (Purified Protein Derivative), results in
localized skin swelling and redness. Therefore, intra-
dermal injection of PPD will reveal whether or not a
person has been infected with Mycobacterium tubercu-
losis. This is important because many infected individ-
uals will not manifest a clinical infection for years.
When a positive PPD test occurs, you can treat and
eradicate the disease before it significantly damages the
lungs or other organs.
When you have a patient with a low-grade fever and
cough, or a patient who has been in contact with people
who have tuberculosis (you, for example, after working
in the hospital), you will decide to "place a PPD." You
inject the PPD intradermally (just barely under the
skin so that the skin bubbles up). Macrophages in the
skin will take up the antigen and deliver it to the T-
cells. The T-cells then move to the skin site, release
lymphokines that activate macrophages, and within
1-2 days the skin will become red, raised, and hard. A
positive test is defined as an area of induration (hard-
ness) that is bigger in diameter than 10 mm after 48
hours (the time it takes for a type IV delayed hyper-
sensitivity reaction to occur). The test is positive at
CHAPTER 14.
MYCOBACTERIUM
104
5 mm of induration in patients who are immunocom-
promised, such as those with AIDS.
Note that a positive test does not mean that the pa-
tient has active tuberculosis; it indicates exposure and
infection to Mycobacterium tuberculosis at some time
in the past. A positive test is present in persons with ac-
tive infection, latent infection, and in those who have
been cured of their infection.
False positive test: You still must be wary with this
test because some people from other countries have had
the BCG (bacillus Calmette-Guerin) vaccine for tu-
berculosis. This vaccine is debatably effective in pre-
venting tuberculosis but it causes a positive PPD.
False negative test: Some patients do not react to
the PPD even if they have been infected with tubercu-
losis. These patients are usually anergic, which
means that they lack a normal immune response due
to steroid use, malnutrition, AIDS, etc. To determine
whether a patient is anergic or just has not been in-
fected with tuberculosis, a second injection (either with
Candida or mumps antigen) is given in the other arm.
Most people have been exposed to these antigens, so
only individuals who are anergic will not respond to
the Candida or mumps injection with induration after
48 hours.
Clinical Manifestations
The first exposure to Mycobacterium tuberculosis is
called primary tuberculosis and usually is a subclini-
cal (asymptomatic) lung infection. Occasionally, an
overt symptomatic primary infection occurs.
When an asymptomatic primary infection occurs, the
acquired cell-mediated immunity will wall off and
suppress the bacteria. These defeated bacteria lie dor-
mant but can later rise up and cause disease. This sec-
ond infection is called secondary or reactivation
tuberculosis.
For the real number crunchers, here are the statistics:
Close contacts, such as household members, of someone
with pulmonary tuberculosis have a 30% chance of being
infected. Of all the infected persons, about 5% will de-
velop tuberculosis in the next 1 or 2 years and 5% will
develop reactivation tuberculosis sometime later in life.
So there is a 10% lifetime risk of developing tuberculosis
for those infected with Mycobacterium tuberculosis.
Primary Tuberculosis
1) Mycobacterium tuberculosis is usually transmit-
ted via aerosolized droplet nuclei from the aerosolized
respiratory secretions of an adult with pulmonary tu-
berculosis. This adult will shower the air with these se-
cretions when he coughs, sings, laughs, or talks.
2) The inspired droplets land in the areas of the lung
that receive the highest air flow: the middle and lower
lung zones. Here there will be a small area of pneu-
momtis with neutrophils and edema, just like any bac-
terial pneumonia.
3) Now the bacteria enter macrophages, multiply,
and spread via the lymphatics and bloodstream to the
regional lymph nodes, other areas of the lungs, and dis-
tant organs.
Tuberculosis is a confusing disease because so many
different things can happen. As cell-mediated immunity
develops, 1) the infection can be contained so that the
patient will not even realize he was infected, or 2) it can
become a symptomatic disease.
1) Asymptomatic primary infection: The cell-
mediated defenses kick in, and the foci of bacteria be-
come walled off in the caseous granulomas. These
granulomas then heal with fibrosis, calcification, and
scar formation. The organisms in these lesions are de-
creased in number but remain viable. Tiny tubercles
(as the granulomas are called) are often too small to be
seen even on chest X-ray. Only a PPD will give the bug-
gers away. Sometimes the chest film will suggest recent
infection by showing hilar lymph node enlargement or
calcifications.
Fig. 14-4.
A calcified tubercle in the middle or lower
lung zone is called a Ghon focus. A Ghon focus accom-
panied by perihilar lymph node calcified granulomas is
called a Ghon, or Ranke, complex.
Figure 14-4
CHAPTER 14.
MYCOBACTERIUM
105
2) Symptomatic primary tuberculosis occurs far
less frequently, more commonly in children, the elderly,
and the immunocompromised (especially HIV infected
persons). These groups do not have as powerful a cell-
mediated immune system as do healthy adults, so the
organisms are not suppressed.
Fig. 14-5.
Overt or manifest primary tuberculosis:
Large caseous granulomas develop in the lungs or other
organs. In the lungs the caseous material eventually
liquifies, is extruded out the bronchi, and leaves behind
cavitary lesions, shown here with fluid in the cavities
(called "cavitary lesions with air-fluid levels" on chest
X-ray).
Secondary or Reactivation Tuberculosis
Most adult cases of tuberculosis occur after the bac-
teria have been dormant for some time. This is called
reactivation or secondary tuberculosis. The in-
fection can occur in any of the organ systems seeded
during the primary infection. It is presumed that a
temporary weakening of the immune system may pre-
cipitate reactivation. Many AIDS patients develop tu-
berculosis in this manner. HIV infected patients who
are infected with
Mycobacterium tuberculosis
have a
10% chance/year of developing reactivation tuberculo-
sis! And
1
/s of HN infected persons are also infected
with
Mycobacterium tuberculosis
(worldwide)!
Figure 14-5
Figure 14-6
Risk of Reactivation in all Persons: 10% for Lifetime!
Risk of reactivation in HIV infected: 10% per year!
Fig. 14-6. The organ systems that can be involved in
tuberculosis:
1) Pulmonary tuberculosis: This is the most com-
mon site of reactivation tuberculosis. The infection usu-
ally occurs in the apical areas of the lung around the
clavicles. It normally reactivates in the upper lobe be-
cause oxygen tension is the highest there, due to de-
creased pulmonary circulation, and Mycobacterium
tuberculosis is an aerobic bacterium. Slowly these areas
of infection grow, caseate, liquify, and cavitate. Clini-
cally, the patients usually present with a chronic low-
grade fever, night sweats, weight loss, and a productive
cough that may have blood in it. This slow erosive in-
fection occurs as the host macrophages and T-cells bat-
tle to wall off the bacteria.
2) Pleural and pericardial infection: Infection in
these spaces results in infected fluid collections around
the lung or heart respectively.
3) Lymph node infection: Worldwide, this is the
most common extrapulmonary manifestation of tuber-
culosis. The cervical lymph nodes are usually involved.
They become swollen, mat together, and drain. Lymph
node tuberculosis is called scrofula.
4) Kidney: Patients will have red and white blood
cells in the urine, but no bacteria are seen by Gram stain
or grow in culture (remember that Mycobacterium tuber-
culosis
takes weeks to grow in culture and are acid-fast).
This is referred to as sterile pyuria.
CHAPTER 14. MYCOBACTERIUM
106
5) Skeletal: This usually involves the thoracic and
lumbar spine, destroying the intervertebral discs and
then the adjacent vertebral bodies (Pott's disease).
6) Joints: There is usually a chronic arthritis of
1 joint.
7) Central nervous system: Tuberculosis causes
subacute meningitis and forms granulomas in the brain.
8) Miliary tuberculosis: Tiny millet-seed-sized tu-
bercles (granulomas) are disseminated all over the body
like a shotgun blast. The kidneys, liver, lungs, and other or-
gans are riddled with the tubercles. A chest film will some-
times show a millet-seed pattern throughout the lung. This
disease usually occurs in the elderly and in children.
BIG PICTURE: Tuberculosis is usually a
chronic disease; it presents slowly with weight
loss, low-grade fever, and symptoms related to
the organ system infected. Because of its slow
course, it may be confused with cancer. When-
ever you have an infection of any organ system,
tuberculosis will be somewhere on your differen-
tial diagnosis list. It is one of the
great imitators!
Diagnosis
1) PPD skin test: This screening test indicates an
exposure sometime in the past.
2) Chest X-ray: You may pick up an isolated gran-
uloma, Ghon focus, Ghon complex, old scarring in the
upper lobes, or active tuberculous pneumonia.
3) Sputum acid-fast stain and culture: When the
acid-fast stain or culture are positive, this indicates an
active pulmonary infection.
The treatment and control of tuberculosis is compli-
cated and will be discussed in the mycobacterial antibi-
otics chapter (see Chapter 18).
Tuberculosis "Rule of Fives"
•
Droplet nuclei are 5 micrometers and contain
5 My-
cobacterium tuberculosis bacilli.
•
Patients infected with Mycobacterium tuberculosis
have a 5% risk of reactivation in the first 2 years and
then a 5% lifetime risk.
P
atients with "high five" NW will have a
5+5%
risk
of reactivation per year!
ATYPICAL MYCOBACTERIA
A large group of mycobacteria live in water and soil
mostly in the southern U. S. Based on tuberculin reac-
tions specific for these organisms (like the PPD), it
has been estimated that up to 50% of the southern
Figure 14-7
population have been infected subclinically. These bac-
teria rarely produce an overt infection, and when they
do, it is usually a pneumonia milder than pulmonary tu-
berculosis, or a skin granuloma or ulcer (see Fig. 14-11).
One particular organism in this group deserves
mention because it has become an important pathogen
in AIDS patients. Mycobacterium avium-intracel-
lulare
( MAI), also called Mycobacterium avium-complex
(MAC), usually only infects birds (avium) and other ani-
mals. It has now become one of the major systemic bac-
terial infections of AIDS patients, usually late in the
course of the disease. In fact, 50% of AIDS patients ex-
amined at autopsy are found to be infected with MAI. It
is rarely the cause of death; however, it is certainly a
harbinger of death as it only strikes when the T-helper
count is virtually nonexistent (see Chapter 25). Infection
with MAI results in a chronic wasting illness; the bacte-
ria disseminate everywhere involving the liver, spleen,
bone marrow, and intestine. The intestinal involvment
often results in chronic watery diarrhea.
Mycobacterium leprae
( Leprosy, also called Hansen's Disease)
Like Mycobacterium tuberculosis, Mycobacterium
leprae
is an acid-fast rod. It is i mpossible to grow this
bacterium on artificial media; it has only been grown in
the footpads of mice, in armadillos, and in monkeys. It
causes the famous disease leprosy.
There are around 6 million persons infected with
Mycobacterium leprae worldwide, with cases focused in
endemic areas such as India, Mexico, Africa, and the
CHAPTER 14. MYCOBACTERIUM
10 7
Pacific Islands (Hawaii included). Every year in the
U.S. there are close to 200 newly diagnosed cases, usu-
ally in immigrants. It is unclear why some people are in-
fected and some are not. Many studies have attempted
to infect human volunteers, with little success. Infection
occurs when a person (who for unknown reasons is sus-
ceptible) is exposed to the respiratory secretions or, less
likely, skin lesions of an infected individual.
The clinical manifestations of leprosy are dependent
on 2 phenomena: 1) The bacteria appear to grow better
in cooler body temperatures closer to the skin surface.
2) The severity of the disease is dependent on the host's
cell-mediated immune response to the bacilli (which
live a facultative intracellular existence, like Mycobac-
terium tuberculosis).
Fig. 14-7.
The acid-fast rod Mycobacterium leprae is
seen here cooling off on an ice cube. Leprosy involves the
cooler areas of the body. It damages the skin (sparing
warm areas such as the armpit, groin, and perineum),
the superficial nerves, eyes, nose and testes.
Cell-mediated immunity once again plays an important
role in the pathogenesis of this disease. The cellular im-
munity that limits the spread of the bacteria also causes
inflammation and granulomas, particularly in skin and
nerves. Clinically, leprosy is broken up into five subdi-
visions based on the level of cell-mediated immunity,
which modulates the severity of the disease:
1) Lepromatous leprosy (LL): This is the severest
form of leprosy because patients canNOT mount a cell-
mediated immune response to Mycobacterium leprae. It
is theorized that defective T-suppressor cells (T-S cells)
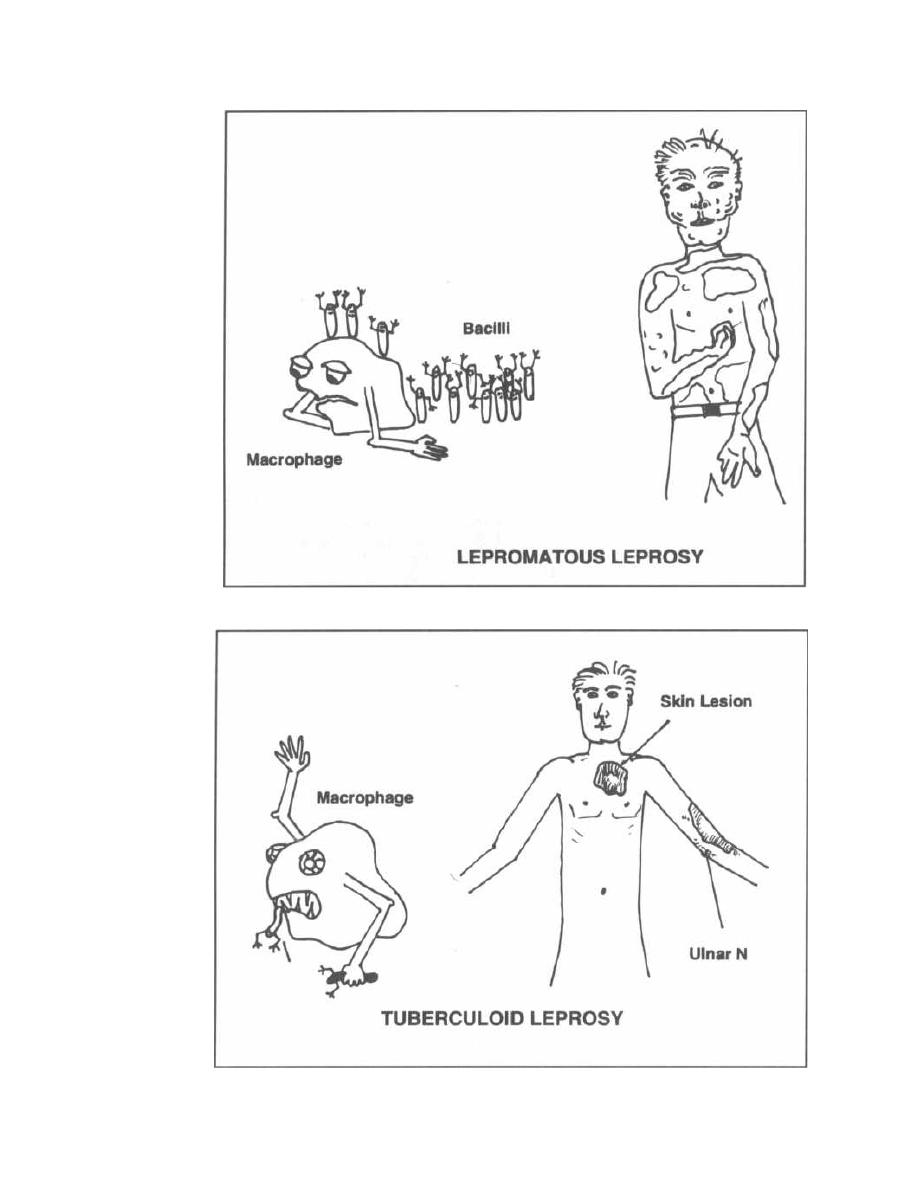
Figure 14-8
CHAPTER 14. MYCOBACTERIUM
Figure 14-9
108

block the T-helper cell's response to the
Mycobacterium
leprae
antigens.
Fig. 14-8.
Lepromatous leprosy (LL): The defeated
macrophage is covered with
Mycobacterium leprae
acid-
fast rods, demonstrating the very low cellular immunity.
The patient with LL cannot mount a delayed hypersensi-
tivity reaction. LL primarily involves the skin, nerves,
eyes and testes, but the acid-fast bacilli are found every-
where (respiratory secretions and every body organ). The
skin lesions cover the body with all sorts of lumps and
thickenings. The facial skin can become so thickened that
the face looks lionlike (hence, leonine facies). The nasal
cartilage can be destroyed, creating a saddlenose de-
formity, and there is internal testicular damage (leading
to infertility). The anterior segment of the eyes can be-
come involved, leading to blindness. Most peripheral
nerves are thickened, and there is loss of sensation in the
extremities in a glove and stocking distribution. The in-
ability to feel in the fingers and toes leads to repetitive
trauma and secondary infections, and ultimately contrac-
tion and resorption of the fingers and toes. Lepromatous
leprosy will eventually lead to death if untreated.
2) Tuberculoid leprosy (TL): Patients with TL
can mount a cell-mediated defense against the bacteria,
thus containing the skin damage so that it is not exces-
sive. They will have milder and sometimes self-limiting
disease.
Fig. 14-9.
Tuberculoid leprosy: The macrophage gob-
bling up the
Mycobacterium leprae
acid-fast rods
demonstrates the high cell-mediated resistance of tu-
berculoid leprosy. The delayed hypersensitivity reaction
CHAPTER 14. MYCOBACTERIUM
109
is intact, so the lepromin skin test is usually positive.
The patient demonstrates localized superficial, uni-
lateral skin and nerve involvement. In this form of lep-
rosy, there are usually only 1 or 2 skin lesions. They are
well-defined, hypopigmented, elevated blotches. The
area within the rash is often hairless with diminished
or absent sensation, and enlarged nerves near the skin
lesions can be palpated. The most frequently enlarged
nerves are those closest to the skin-the greater auric-
ular, the ulnar (above the elbow), the posterior tibial,
and the peroneal (over the fibula head). The bacilli are
difficult to find in the lesions or blood. Patients are non-
infectious and often spontaneously recover.
The 3 remaining categories represent a continuum
between LL and TL. They are called borderline lep-
romatous (BL), borderline (BB), and borderline tu-
berculoid (BT). The skin lesions of BL will be more
numerous and have a greater diversity of shape than
those of BT.
The lepromin skin test is similar to the PPD used
in tuberculosis. It measures the ability of the host to
mount a delayed hypersensitivity reaction against anti-
gens of
Mycobacterium leprae.
This test is more prog-
nostic than diagnostic and is used to place patients on
the immunologic spectrum. It makes sense that TL pa-
tients would have a positive cell-mediated immune re-
sponse and thus a positive lepromin skin test, while LL
patients, who cannot mount a cell-mediated immune re-
sponse, have a negative response to lepromin.
See Chapter 18 for information about the treatment
of leprosy.
Fig. 14-10.
The spectrum of leprosy.
Fig. 14-11.
Summary of acid-fast bacteria.
(But a glove and stocking peripheral neuropathy, causing hand and feet numb-
ness, is present!)
Adapted from American Medical Association Drug evaluations,
6th edition, p. 1547.
Figure 14-10
SPECTRUM OF LEPROSY
Tuberculoid Borderline
Lepromatous
Number of
skin lesions
Single
Several
Many
Hair growth
on skin
lesions
Absent
Slightly
decreased
Not affected
Sensation in
lesions of the
extremities
Completely
lost
Moderately
lost
Not affected'
Acid fast
bacilli in skin
scrapings
None
Several
I nnumerable
Lepromin
skin test
Strongly
positive
No reaction No reaction
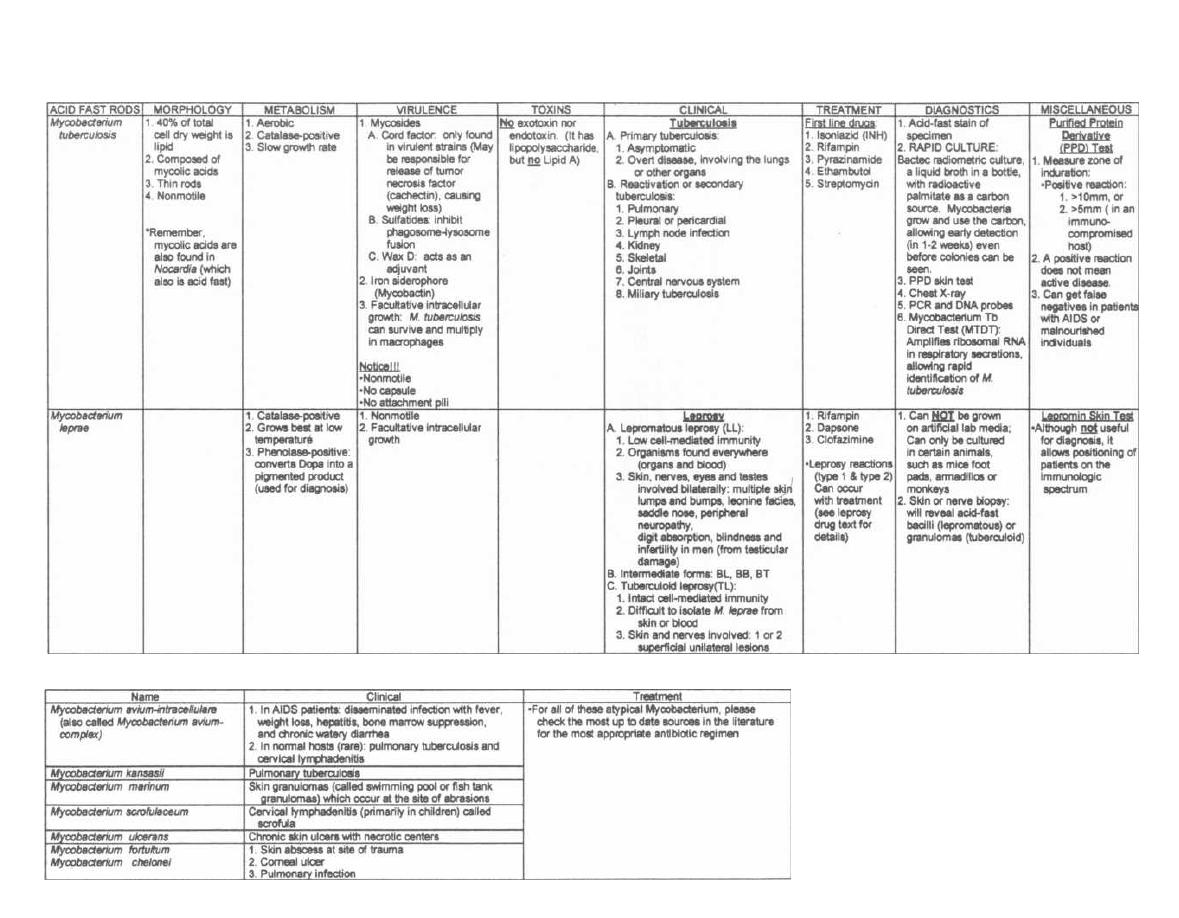
Atypical
Mycobacterium
Figure 14-11
ACID FAST BACTERIA
M. Gladwin and 6. Trattler, Clinical Microbiology Made Ridiculously Simple ®MedMaster

BACTERIA WITHOUT CELL WALLS
The Mycoplasmataceae are the tiniest free-living or-
ganisms capable of self-replication. They are smaller
than some of the larger viruses. Mycoplasmataceae are
unique bacteria because they lack a peptidoglycan cell
wall. Their only protective layer is a cell membrane,
which is packed with sterols (like cholesterol) to help
shield their cell organelles from the exterior environ-
ment. Due to the lack of a rigid cell wall, Mycoplasmat-aceae can contort into a broad range of shapes, from
round to oblong. They therefore cannot be classified as
rods or cocci.
The lack of a cell wall explains the ineffectiveness of
antibiotics
that
attack the cell wall (penicillin,
cephalosporin), as well as the effectiveness of the anti-
ribosomal antibiotics erythromycin and tetracycline.
There are 2 pathogenic species of Mycoplasmat-
aceae,
Mycoplasma pneumoniae
and
Ureaplasma
urealyticum.
Fig. 15- 1.
Mycoplasmataceae surrounded only by a
cell membrane, padded with sterols. Penicillin and
cephalosporin fail to tear down the cell membrane,
while they successfully destroy the cell wall of a nearby
gram-positive Streptococcus.
Mycoplasma pneumoniae
Mycoplasma pneumoniae causes a mild, self-limited
bronchitis and pneumonia. It is the number one cause
of bacterial bronchitis and pneumonia in teenagers and
young adults. Following transmission via the respira-
tory route, this organism attaches to respiratory ep-
ithelial cells with the help of protein P1 (an adhesin
virulence factor). After a 2-3 week incubation period,
infected patients will have a gradual onset of fever, sore
throat, malaise, and a persistent dry hacking cough.
This is referred to as walking pneumonia, because
clinically these patients do not feel very sick.
Chest X-ray reveals a streaky infiltrate, which usu-
ally looks worse than the clinical symptoms and physi-
cal exam suggest. Most symptoms resolve in a week,
although the cough and infiltration (as seen on X-ray)
may last up to 2 months. Although Mycoplasma is a bac-
terium, the nonproductive cough and the streaky infil-
trate on the chest X-ray are more consistent with a viral
(atypical) pneumonia (see Chapter 12, page 83).
Diagnostic tests include:
CHAPTER 15.
MYCOPLASMA
111
1) Cold agglutinins: Certain antigens present on
human red blood cells are identical to antigens of the
Mycoplasma pneumoniae membrane glycolipids. Anti-
bodies to these Mycoplasma pneumoniae antigens cross-
react
with human red blood cell antigens and
agglutinate the red blood cells at 4°C. These antibodies
are thus called cold agglutinins. They develop by the
first or second week of the Mycoplasma pneumoniae in-
fection, peak 3 weeks after the onset of the illness, and
slowly decline over a few months.
You can perform this simple test at the bedside. Put
the patient's blood in a nonclotting tube. After placing
this tube on ice, the blood will clump together if the pa-
tient has developed the cold agglutinin antibodies.
Amazingly, when you lift the tube out of the ice, the
clumped blood will unclump as it warms in the palm of
your hand.
2) Complement fixation test: The patient's serum
is mixed with glycolipid antigens prepared from
My-
coplasma. A fourfold rise in antibody titer between
acute and convalescent samples is diagnostic of a recent
infection.
3) Sputum culture: Mycoplasmataceae (both
M.
pneumoniae and U. urealyticum) can be grown on arti-
ficial media. These media must be rich in cholesterol
and contain nucleic acids (purines and pyrimidines).
After 2-3 weeks, a tiny dome-shaped colony of
My-
coplasma will assume a "fried-egg" appearance.
4) Mycoplasma DNA probe: Sputum samples are
mixed with a labeled recombinant DNA sequence ho-
mologous to that of the mycoplasma. The recombinant
probe will label mycoplasma DNA if present.
This is a self-limiting pneumonia, but erythromycin
and tetracycline will shorten the course of the illness.
Ureaplasma urealyticum
( T-strain Mycoplasma)
Hold on!!! Why isn't this second species of Mycoplas-
mataceae called "Mycoplasma"? The man who named
this tiny organism didn't want you to ever forget that
Ureaplasma loves swimming in urine and produces
urease to break down urea (so it is "urea-lytic"!). It is
sometimes referred to as a T-strain Mycoplasma, as it
produces Tiny colonies when cultured.
Ureaplasma urealyticum is part of the normal flora
in 60% of healthy sexually active women and commonly
STREPTOCOCCAL
PEPTIDOGLYCAN
CELL WALL
C
o
Mycoplasma pneumoniae
0
h'
STEROL PACKED
CELL MEMBRANE
PENICILLIN
PENICILLIN
Figure 15-1
infects the lower urinary tract, causing urethritis. Ure-
Ureaplasma urealyticum can be identified by its abil-
thritis is characterized by burning on urination (dy-
ity to metabolize urea into ammonia and carbon dioxide.
curia) and sometimes a yellow mucoid discharge from
the urethra. Neisseria gonorrhoeae and Chlamydia tra-
Fig. 15-2. Summary of the Mycoplasmataceae.
chomatis are the other 2 bacteria that cause urethritis
(see Chapter 12, page 80).
CHAPTER 15. MYCOPLASMA
112
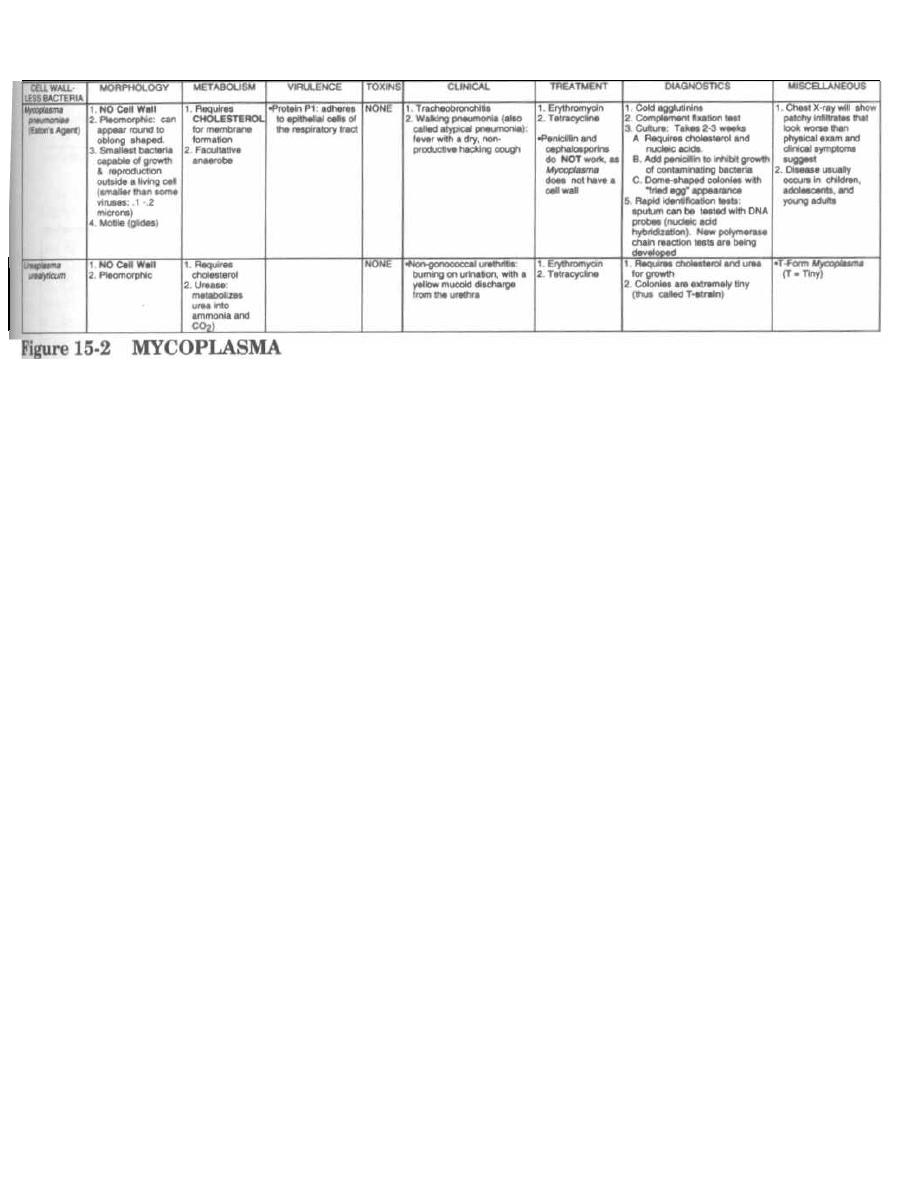
I
CHAPTER 15.
MYCOPLASMA
113
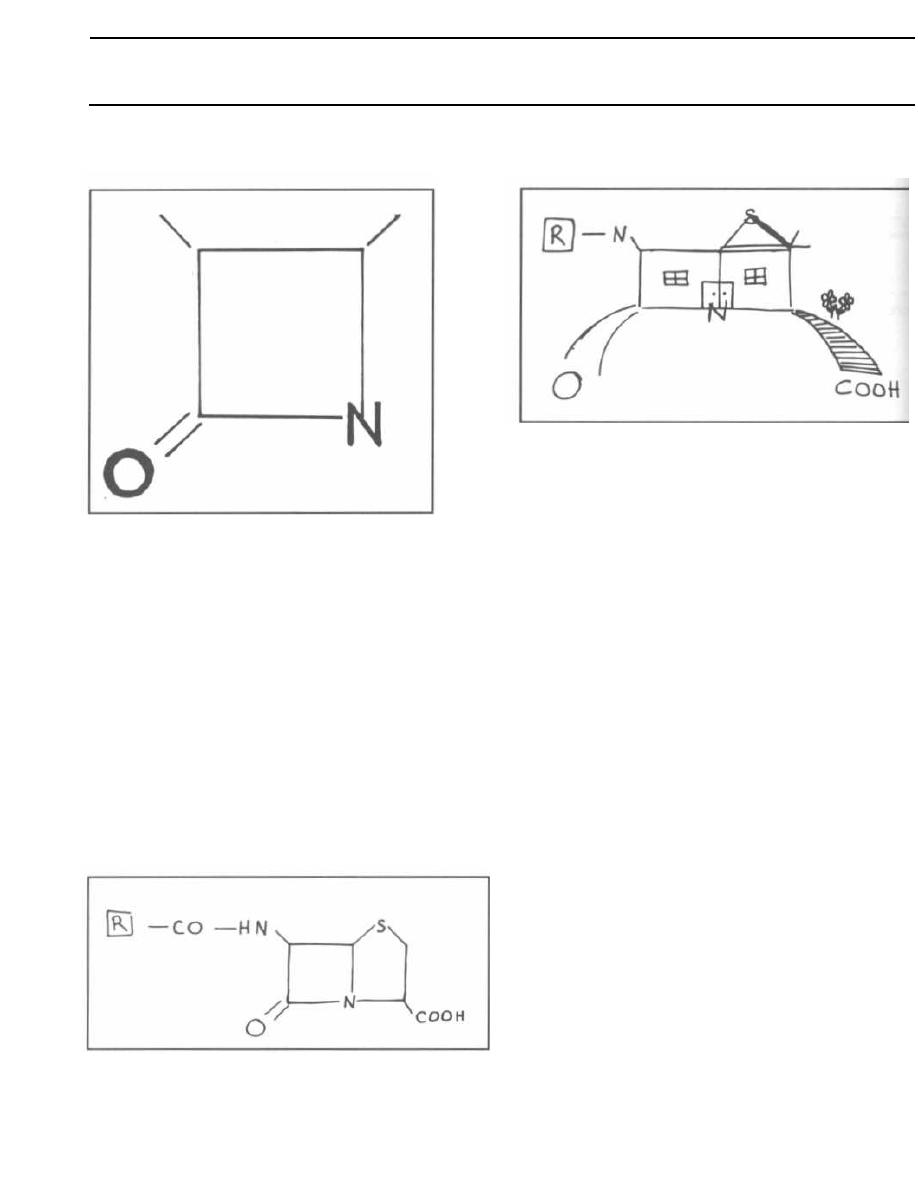
Figure 16-1
Since its introduction during World War II, penicillin
has provided a safe and effective treatment for a multi-
tude of infections. Over time, many bacteria have de-
signed ways to defeat penicillin. Fortunately, scientists
have continued to develop new types of penicillins, as
well as other antibiotics that are able to overcome most
of the bacterial defenses.
Fig. 16-1. This simple-looking box is a beta-lactam
ring. All penicillin-family antibiotics have a beta-lactam
ring. For this reason they are also called the beta-
lactam antibiotics.
Fig. 16-2. Penicillin has another ring fused to the
beta-lactam ring.
ANTI-BACTERIAL MEDICATIONS
CHAPTER 16. PENICILLIN-FAMILY ANTIBIOTICS
Figure 16-2
114
Figure 16-3
Fig. 16-3. Penicillin home. This looks like a house with
a new room built on the side. Notice the funky antenna
that run the groovy sound system. You will see later that
changing the antennae, adding another antenna, or
building a basement will create new types of penicillin
with differing spectrums of activity and potencies.
Mechanism of Action
The penicillins don't just slow the growth of bacteria,
they kill bacteria. They are therefore bacteriacidal.
You will recall (see Chapter 1) that both gram-posi.
tive and gram-negative bacteria possess peptidoglycans
in their cell walls. These are composed of repeating dis-
accharide units cross-linked with amino-acids (pep-
tides). The enzyme that catalyzes this linkage is called
a transpeptidase.
The penicillin must evade the bacterial defenses and
penetrate the outer cell-wall layers to the inner cytoplas.
mic membrane, where the transpeptidase enzymes are
located. In gram-negative bugs, the penicillin must pass
through channels known as porins. Then the penicillin
beta-lactam ring binds to and competitively in-
hibits the transpeptidase enzyme. Cell wall synthe-
sis is arrested, and the bacteria die. Because penicillin
binds to transpeptidase, this enzyme is also called the
penicillin-binding protein.
To be effective the beta-lactam penicillin must:
1) Penetrate the cell layers.
2) Keep its beta-lactam ring intact.
3) Bind to the transpeptidase (penicillin-binding protein).
Resistance to Beta-Lactam Antibiotics
Bacteria defend themselves from the penicillin fam-
ily in 3 ways. Gram-positive bacteria and gram-negative
bacteria use different mechanisms:
1) One way that gram-negative bacteria defend
themselves is by preventing the penicillin from pene-
trating the cell layers by altering the porins. Remem-
ber that gram-negative bacteria have an outer lipid
bilayer around their peptidoglycan layer (see Chapter
1). The antibiotic must be the right size and charge to
be able to sneak through the porin channels, and some
penicillins cannot pass through this layer. Because
gram-positive bacteria do not have this perimeter de-
fense, this is not a defense that gram-positives use.
2) Both gram-positive and gram-negative bacteria
can have beta-lactamase enzymes that cleave the C-N
bond in the beta-lactam ring.
Figure 16-4
Fig. 16-4.
Beta-lactamase enzyme (depicted here as a
cannon) cleaves the C-N bond.
Gram-positive bacteria (like Staphylococcus aureus)
secrete the beta-lactamase (called penicillinase in
the secreted form) and thus try to intercept the antibi-
otic outside the peptidoglycan wall.
Gram-negative bacteria, which have beta-lactamase
enzymes bound to their cytoplasmic membranes, de-
stroy the beta-lactam penicillins locally in the periplas-
mic space.
3) Bacteria can alter the molecular structure of the
transpeptidase so that the beta-lactam antibiotic will
not be able to bind. Methicillin-resistant Staphylococcus
aureus (MRSA) defends itself in this way, making it re-
sistant to ALL of the penicillin family drugs.
CHAPTER 16. PENICILLIN-FAMILY ANTIBIOTICS
115
Adverse Effects
All penicillins can cause anaphylactic (aller-
gic) reactions. An acute allergic reaction may occur
from minutes to hours and is IgE-mediated. Bron-
chospasm, urticaria (hives), and anaphylactic shock
(loss of ability to maintain blood pressure) can occur.
More commonly, a delayed rash appears several days
to weeks later.
All of the penicillin family antibiotics can cause diar-
rhea by destroying the natural GI flora and allowing re-
sistant
pathogenic bacteria (such as
Clostridium
dificile ) to grow in their place.
Types of Penicillin
There are 5 types:
1) Penicillin G: This is the original penicillin dis-
covered by Fleming, who noted that the mold Penicil-
lium notatum produced a chemical that inhibited
Staphylococcus aureus. Penicillin was first used in hu-
mans in 1941.
2) Aminopenicillins: These penicillins offer better
coverage of gram-negative bacteria.
3) Penicillinase-resistant penicillins: This group is
useful against beta-lactamase (an enzyme that destroys
beta-lactam rings) producing Staphylococcus aureus.
4) Anti-Pseudomonal penicillins (including the
carboxypemcillins, ureidopenicillins, and monobactams):
This group offers even wider coverage against gram-
negative bacteria (including Pseudomonas aeruginosa).
5) Cephalosporins: This is a widely used group of
antibiotics that have a beta-lactam ring, are resistant to
beta-lactamase, and cover a broad spectrum of gram-
positive and gram-negative bacteria.
Many bacteria produce cephalosporinases, making
them resistant to many of these drugs.
Penicillin G
Fig. 16-5.
Penicillin G is the original G-man of the
penicillins. There are oral dosage formulations of Peni-
cillin G, but it is usually given intramuscularly (IM) or
i ntravenously TV. It is usually given in a crystalline
form to increase i ts half-life.
Many organisms have now developed resistance to
the old G-man because he is sensitive to beta-lactamase
enzymes. But there are a few notable times when the G-
man is still used:
1) Pneumonia caused by Streptococcus pneumoniae.
( However, resistant strains are developing.)
Penicillin V is an oral form of penicillin. It is
acid stable in the stomach. It is commonly given for
Figure 16-5
streptococcus pharyngitis caused by group A beta-he-
molytic streptococcus since it can be taken orally.
Aminopenicillins
(Ampicillin and Amoxicillin)
These drugs have a broader spectrum than Peni-
cillin G, hitting more gram-negative organisms. This
enhanced gram-negative killing is attributable to better
penetration through the outer membranes of gram-neg-
ative bacteria and better binding to the transpeptidase.
However, like penicillin G, the aminopenicillins are still
inhibited by penicillinase.
The gram-negative bacteria killed by these drugs in-
clude
Escherichia coli
and the other enterics
(Proteus,
Salmonella, Shigella,
etc.). However, resistance has de-
veloped: 30% of
Haemophilus influenzae
and many of
the enteric gram-negative bacteria have acquired peni-
cillinase and are resistant.
CHAPTER 16. PENICILLIN-FAMILY ANTIBIOTICS
11 6
Note that the aminopenicillins are one of the few
drugs effective against the gram-positive enterococcus
(see Fig. 16-17).
Both ampicillin and amoxicillin can be taken orally,
but amoxicillin is more effectively absorbed orally so
you will frequently use it for outpatient treatment of
bronchitis, urinary tract infections, and sinusitis,
caused by gram-negative bacteria.
IV ampicillin is commonly used with other antibiotics
such as the aminoglycosides (gentamicin) for broad
gram-negative coverage. In the hospital you will become
very familiar with the "Amp-gent" combo! Patients
with serious urinary tract infections are often infected
with a gram-negative enteric or enterococcus. Amp-gent
offers a perfect broad empiric coverage until cultures re-
veal the exact organism responsible.
Figure 16-6
Penicillinase-Resistant Penicillins
Methicillin, nafcillin, and oxacillin are penicilli-
nase-resistant drugs that can kill
Staphylococcus au-
reus.
These are usually given IV.
Methicillin was highly efficacious against staphylo-
coccal infections, but because of the occurrence of inter-
stitial nephritis, its use has been discontinued in the
United States. You will still hear its name used fre-
quently in reference to sensitivity testing (e.g. Methi-
cillin Resistant
Staphylococcus aureus).
Fig. 16-6. This picture will help you remember the
names of the IV beta-lactamase resistant penicillins:
I met a nasty ox with a beta-lactamase ring around
its neck.
Nafcillin is the drug of choice for serious
Staphylo-
coccus aureus
infections, such as cellulitis, endocarditis,
and sepsis.
Figure 16-7. THE
CLOX
WERE TICKING.
Fig. 16-7.
The clocks ( clox) were ticking. It was only
a matter of time before the oral beta-lactamase re-
sistant penicillins were discovered: Cloxacillin and di-
cloxacillin.
There are now oral formulations of nafcillin and
oxacillin.
These drugs are not good against gram-negative or-
ganisms. They are used for gram-positive bacteria,
especially those that produce penicillinase ( Staphylo-
coccus aureus).
When a patient has an infected skin wound (cellulitis,
i mpetigo, etc.), you know he most likely has Staphylo-
coccus aureus or group A beta-hemolytic streptococcus.
Treating with Penicillin G, V, or ampicillin would not
cover penicillinase-producing Staphylococcus aureus.
Treating with one of these penicillinase-resistant agents
will, and if you give him one of the oral agents he can go
home on oral antibiotics. You won't have to take care of
him around the clock!!!
Anti-Pseudomonal Penicillins
( Carboxypenicillins and Ureidopenicillins)
This group of penicillins has expanded gram-
negative rod coverage, especially against the difficult-
to-destroy Pseudomonas aeruginosa. They are also ac-
tive against anaerobes (Bacteroides fragilis) and many
gram positives.
CHAPTER 16. PENICILLIN-FAMILY ANTIBIOTICS
11 7
Fig. 16-8.
Pseudomonas, which can cause a devastat-
ing pneumonia and sepsis, is resistant to many antibi-
otics. It is so crafty and sneaky that we need James
Bond to help with its elimination. Bond is fortunate to
have three excellent weapons for his task. He has his
pick of a car ( with special weapons and gadgets), a spe-
cially trained tick that can home in on its target and
suck out the life of the target, or a megaton pipe bomb:
Carboxypenicillins: Ticarcillin
and Carbenicillin
Ureidopenicillins: Piperacillin and mezlocillin.
Like ampicillin, these drugs are combined with an
aminoglycoside to double up the Pseudomonas killing
(synergism). Frequently used combos include "Pip and
gent" and "ticar and gent."
These drugs are sensitive to penicillinases, and thus
most Staphylococcus aureus are resistant. Carbenicillin
has certain disadvantages such as lower activity and
thus the need for high dosages; high sodium load;
platelet dysfunction; and hypokalemia. The parenteral
form is currently not available for use in the United
States. Replacement with ticarcillin or a ureidopeni-
cillin has reduced these problems and provided antibac-
terial activity.
Beta-Lactamase Inhibitors
( Clavulanic Acid, Sulbactam, and Tazobactam)
These enzymes are inhibitors of beta-lactamase. They
can be given in combination with penicillins to create a
beta-lactamase resistant combination:
Amoxicillin and clavulanic acid = Augmentin
(trade name)
Ticaricillin and clavulanic acid = Timentin
(trade name)
Ampicillin and sulbactam = Unasyn (trade name)
Piperacillin and tazobactam = Zosyn (trade name)
These drugs provide broad coverage against the beta-
lactamase producing gram-positives ( Staphylococcus
aureus), gram-negatives (Haemophilus influenza), and
anaerobes ( Bacteroides fragilis).
The Cephalosporins
There are now more than 20 different kinds of
cephalosporins. How do you become familiar with so
many antibiotics? Do not fear. This chapter will teach
you how to master these drugs!
Fig. 16-9.
The cephalosporins have 2 advantages over
the penicillins:
1) The addition of a new basement makes the beta-
lactam ring much more resistant to beta-lactamases
(but now susceptible to cephalosparinases!).
2) A new R-group side chain (another antenna-cable
TV if you will) allows for double the manipulations in
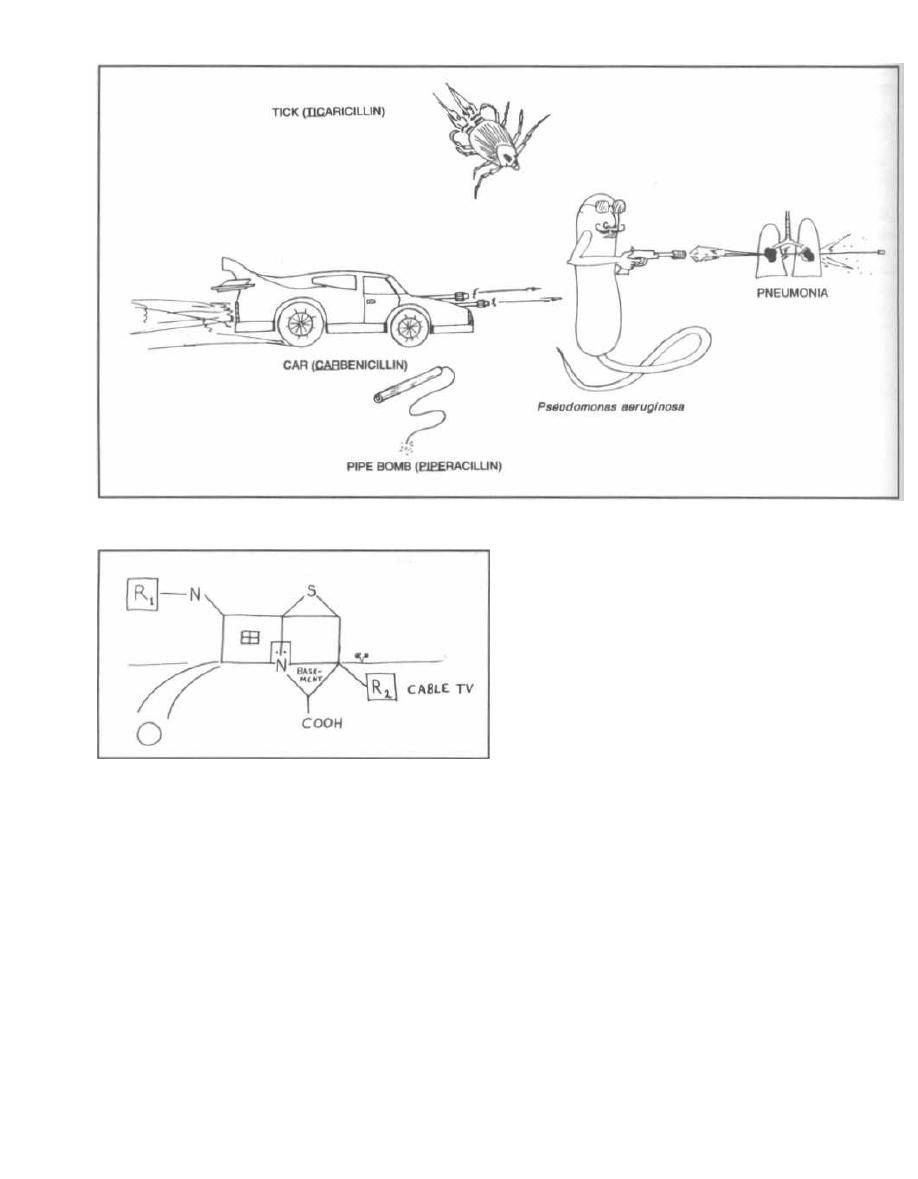
CHAPTER 16. PENICILLIN-FAMILY ANTIBIOTICS
Figure 16-8
Figure 16-9
the lab. This leads to all kinds of drugs with different
spectrums of activity.
There are 3 major generations of cephalosporins:
first, second, and third. These divisions are based on
their activity against gram-negative and gram-positive
organisms.
Fig.
16-10.
With
each
new
generation
of
cephalosporins, the drugs are able to kill an increasing
spectrum of gram negative-bacteria.
At the same time, the newer cephalosporins are less
effective against the gram-positive organisms. The
Streptococci and Staphylococci are most susceptible to
first-generation cephalosporins.
11 8
Note that MRSA (Methicillin Resistant Staphylo-
coccus aureus) is resistant to all cephalosporins be-
cause it has changed the structure of its penicillin
binding protein (transpeptidase). The Enterococci (in.
cluding Streptococcus faecalis) are also resistant to
cephalosporins .
Fig. 16-11.
MRSA and the Enterococci are resistant
to the cephalosporins.
A new cephalosporin has been classified as a
fourth.
generation
antibiotic because it has great gram-negative
coverage like the 3rd generation but also has very good
gram-positive coverage.
The
Names!
How do you remember which
cephalosporin is in which group!!??!! The most impor-
tant thing is to remember the trends and then you can
look in any pocket reference book for the specific drugs
in each group. However, it is nice to be familiar with the
names of individual drugs, and exams often expect you
to be able to recognize them. Here is an easy, although
i mperfect, way to learn many of them.
First-Generation
Almost all cephalosporins have the sound cef in their
names, but the first generation cephalosporins are the
only ones with a PH. To know the first-generation cephalo.
sporins, you first must get a PH.D. in PHarmacology.
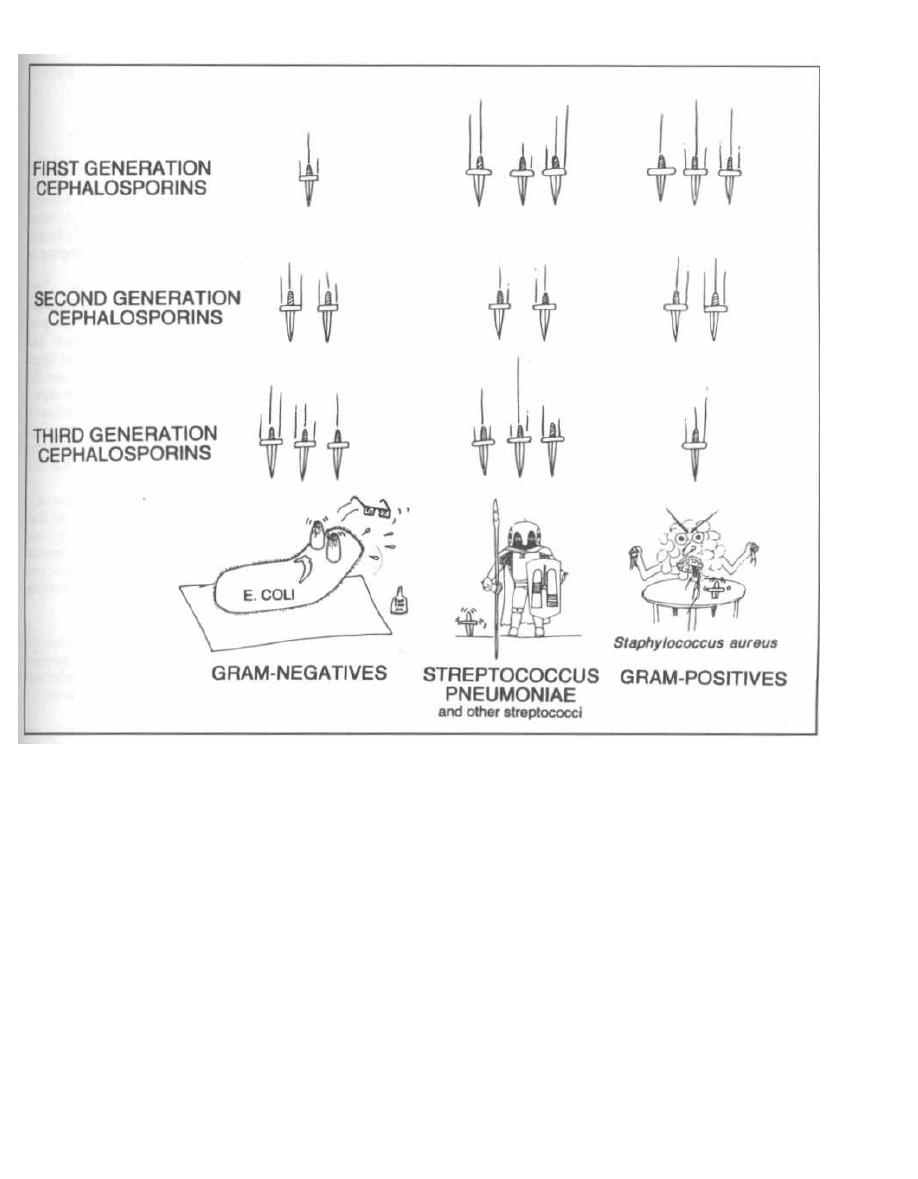
Figure 16-10
cephalothin
cephapirin
( Exceptions: cefazolin, cefadroxil)
cephradine
cephalexin
Cefazolin is an important first-generation drug that
doesn't have a PH. Don't let this faze you!
CHAPTER 16. PENICILLIN-FAMILY ANTIBIOTICS
119
coats, and your FOXY cousin is drinking TEA in a toast
to your achievement.
cefamandole
cefaclor
cefuroxime
( Exceptions:
cefmetazole, cefonicid,
cefprozil, loracarbef)
cefoxitin
cefotetan (pronounced ce-fo-tea-tan)
Second-Generation
Third-Generation
Fig. 16-12.
Second-generation cephalosporins have
fam, fa, fur, fox, or tea, in their names. After you get
TRI for third (you know, triglycerides, etc.). Most of
your PH.D., you would want to gather your family to
the third-generation cephalosporins have a T (for tri) in
celebrate! The FAMily is gathered, some wearing FUR
their names.

Figure 16-11
Figure 16-12
ceftriaxone
ceftazidime
( Exceptions: cefixime, cefoperazone,
cefpodoxin, cefetamet)
cefotaxime
ceftizoxime
ceftibuten
Note that cefotetan (tea) is a second generation drug.
Fourth Generation
There is only one:
cefepime
and it is the only cephalosporin with a fep in its name.
( Cefpirome is an investigational agent that will also
belong in this class.)
Adverse Effects
Ten percent of patients who have allergic reactions to
penicillin will also have a reaction to cephalosporins.
Such allergic reactions are the same as with penicillin:
CHAPTER 16. PENICILLIN-FAMILY ANTIBIOTICS
12 0
an acute IgE-mediated reaction or the more comm
rash, which usually appears weeks later.
"When do we use these antibiotics?"
1) First-generation cephalosporins: Recall Fig,
16-10 showing the excellent gram-positive coverage.
First-generation cephalosporins are used as alterna-
tives to penicillin for staphylococcal and streptococcal.
i nfections when penicillin cannot be tolerated (allergy)..
Surgeons love to give these drugs before surgery
prevent infection from the skin.
2) Second-generation
cephalosporins:
This
group covers more of the gram-negative rods than the
first-generation cephalosporins. Cefuroxime has good
coverage against both
Streptococcus
pneumoniae and
Haemophilus influenzae. This makes it an ideal agent
for community-acquired bacterial pneumonia when the
sputum is negative and you don't know what the organ-
ism is.
( Streptococcus pneumoniae
and Haemophilus
in.
fluenza are common causes of community-acquired
pneumonia.) Cefuroxime is also good for sinusitis and
otitis media, which are often caused
by
Haemophilus in-
fluenza or Branhamella
catarrhalis.
Anaerobic
coverage:
Three
second-generation
cephalosporins cover anaerobic bacteria, such as
Bac
teroides fragilis.
These can be used for intra-abdominal
i nfections,
aspiration
pneumonias, and colorectal
surgery prophylaxis, all of which involve anaerobic con-
tamination from the GI tract. These 3 drugs are cefote-
tan, cefoxitin, and cefmetazole
Fig. 16-13.
Some of the second-generation cephalo-
sporins kill anaerobic bacteria, such as Bacteroides
fragilis.
Study the picture of a fox (cefoxitin) who met
(cefmetazole) an anaerobic bug for tea (cefotetan).
3) Third-generation cephalosporins: These are
used for the multi-drug resistant aerobic gram-negative or-
ganisms that cause nosocomial (hospital-acquired) pneu-
monia, meningitis, sepsis, and urinary tract infections.
The fourth generation cefepime is sometimes called an
extended spectrum 3rd-generation cephalosporin. Think
of him as the same but with a little added muscle against
gram-positives and the terrible Pseudomonas aeruginosa
Ceftazidime, cefoperazone, and cefepime are
the only cephalosporins that are effective against
Pseudomonas aeruginosa. So when you encounter the
"impossible-to-kill" Pseudomonas: Give it the Taz, the
Fop, and the Fep!
Ceftriaxone has the best CSF penetration and covers
the bacteria that frequently cause meningitis. It is the
first-line drug for meningitis in neonates, children, and
adults. Ceftriaxone is also given IM for gonorrhea, as
more Neisseria gonorrhoea have become resistant to
penicillin and tetracycline.

CHAPTER 16. PENICILLIN-FAMILY ANTIBIOTICS
Figure 16-13
Figure 16-14
121

Figure 16-15
ANTIBIOTICS
THAT COVER PSEUDOMONAS
AER UGINOSA
Imipenem
There is now a new class of beta-lactam antibiotics called
the carbapenems. You need to know one of its members...
Tell yourself that you are a pen. Read: "I'm a pen."
Now picture the pen crossing out all the bacteria that
are difficult to treat. The pen (imipenem) can terminate
almost all of them.
Imipenem has the broadest antibacterial activ-
ity of any antibiotic known to man!!! It kills gram-
negatives, gram-positives, and anaerobes (even tough
guys like
Pseudomonas aeruginosa
and Enterococcus).
Some bacteria that are still resistant to this drug include
our enemy MRSA, some
Pseudomonas
species, and bac-
teria without peptidoglycan cell walls
(Mycoplasma).
Imipenem is stable to beta-lactamases. Because it is
very small, it can pass through porin channels to the
periplasmic space. There it can interact with transpep-
tidase in a similar fashion as the penicillins and
cephalosporins. Unfortunately, with heavy use of this
antibiotic some bacterial strains have developed new
enzymes that can hydrolyze imipenem, and some gram-
negative bacteria have squeezed down their porin chan-
nels to prevent its penetration.
The normal kidney has a dihydropeptidase that
breaks imipenem down, so a selective enzyme inhibitor
of this dihydropeptidase is given with imipenem. The
inhibitor is cilastin.
Imipenem can cause allergic reactions similar to those
of penicillin. This drug also lowers the seizure threshold.
CHAPTER 1 6. PENICILLIN-FAMILY ANTIBIOTICS
122
Figure 16-16
ANTIBIOTICS
THAT COVER THE ANAEROBES
(INCLUDING BACTEROIDES
FRAGILIS)
Clinical note: On the wards imipenem is called
a "decerebrate antibiotic" because you don't have to
think about what bacteria it covers. It covers almost
everything!!!
Meropenem is a newer carbapenem that is as
powerful as imipenem. It can be used interchangeably.
Meropenem is stable against dihydropeptidase, so
cilastin is not needed. Meropenem also has a reduced
potential for causing seizures in comparison with
Imipenem.
Aztreonam
Aztreonam is a magic bullet for gram-negative
aerobic bacteria!!! It is a beta-lactam antibiotic, but it
is different in that it is a monobactam. It only has the
beta-lactam ring, with side groups attached to the ring.
It does not bind to the transpeptidases of gram-positive
or anaerobic bacteria, only to the transpeptidase of
gram-negative bacteria.
Fig. 16-14 A TREE (AzTREonam) has fallen through
the center of our house, leaving only the square portion
(beta-lactam ring) standing, and letting all the air in
(aerobic). You can imagine if this happened to your
house it would be a negative (gram) experience. Aztre-
onam kills gram-negative aerobic bacteria.
Aztreonam kills the tough hospital-acquired, multi-
drug resistant, gram-negative bacteria, including
Pseudomonas aeruginosa.
Data suggest there is little cross-reactivity with the
bicyclic beta-lactams, so we can use this in penicillin-al-
lergic patients!
Clinical notes: Because this antibiotic only kills
gram-negative bugs, it is used (much like the aminogly-
1. Antipseudomonal penicillins
A. Ticarcillin
B. Timentin (ticarcillin & clavulanate)
C. Piperacillin
D. Zosyn (piperacillin & tazobactam)
E. Carbenicillin
F. Mezlocillin
2. Third generation cephalosporins
A. Ceftazidime
B. Ceftizoxime
C. Cefoperazone
3. Imipenem
4. Aztreonam
5. Quinolones
A. Ciprofloxacin
B. Levofloxacin
C. Trovafloxacin
6. Aminoglycosides
A. Gentamicin
B. Tobramycin
C. Amikacin
1. Penicillins with beta-lactamase inhibitor
A. Augmentin (Amoxicillin & clavulanate)
B. Timentin (Ticarcillin & clavulanate)
C. Unasyn (Ampicillin & sulbactam)
D. Zosyn (Pipericillin & tazobactam)
2. Second generation cephalosporins
A. Cefoxitin
B. Cefotetan
C. Cefmetazole
3. Imipenem and Meropenem
4. Chloramphenicol
5. Clindamycin
6. Metronidazole
7. Trovafloxacin`

CHAPTER 1 6. PENICILLIN-FAMILY ANTIBIOTICS
Figure 16-17 ANTIBIOTICS THAT COVER THE DIFFICULT-
TO-KILL GRAM-POSITIVE BACTERIA
cosides) along with an antibiotic that covers gram-posi-
tives. The resulting combinations give powerful broad-
spectrum coverage:
vancomycin + aztreonam
clindamycin + aztreonam
Fig.
16-15.
Antibiotics that cover Pseudomonas
aeruginosa.
Fig. 16-16.
Antibiotics that cover anaerobic bacteria,
including Bacteroides fragilis.
Fig. 16-17.
Antibiotics that cover the difficult-to-kill
gram-positive bacteria: methicillin-resistant Staphylo-
coccus aureus (MRSA), the enterococci, and methicillin-
resistant Staphylococcus epidermidis.
Fig. 16-18.
Summary of the penicillin (beta-lactam)
family antibiotics.
References
Fish DN, Singletary TJ. Meropenem, a new carbapenem an-
tibiotic. Pharmacotherapy 1997; 17:644-669.
Fraser KL, Grossman RF. What new antibiotics to offer in the
outpatient setting. Sem Resp Infect 1998; 13:24-35.
Gilbert DN, Moellering RC, Sande MA. The Sanford Guide to
Antimicrobial Therapy 1998. 28th edition. Antimicrobial
Therapy Inc. Dallas Texas, 1998.
Mandell GL, Bennett JE, Dollin R, eds. Principles and Prac-
tice of Infectious Diseases; 4th edition. New York: Living-
stone 1995.
Owens RC, Nightingale CH, et al. Ceftibuten: An overview.
Pharmacotherapy 1997; 17:707-720.
Rockefeller University Workshop. Special report: multiple-
antibiotic-resistant pathogenic bacteria. N Engl J Med
1994;330:1247-1251.
123
Recommended Review Articles:
Hellinger WC, Brewer NS. Carbapenems and Monobactams:
Imipenem, Meropenem, and Aztreonam. Symposium on anti-
microbial agents. Mayo Clinic Proc 1999;74:420-434.
Marshall WF, Blair JE. The cephalosporins. Symposium on
antimicrobial agents. Mayo Clin Proc 1999;74:187-195.
Wright, AJ. The penicillins. Symposium on antimicrobial
agents. Mayo Clinic Proc 1999;74:290-307.
*Because of liver toxicity, the FDA advised that
trovafloxacin should be reserved for treatment
ONLY in patients that meet ALL of the following
criteria:
Who have been at least one of five types of serious and
life threatening infections listed below that is judged by
the treating physician to be serious and life or limb-
threatening:
•
Nosocomial pneumonia (pneumonia acquired in the
hospital)
•
Community acquired pneumonia
•
Complicated intra-abdominal infections, including
post-surgical infections
•
Gynecological and pelvic infections
•
Complicated skin and skin structure infections, in-
cluding diabetic foot infections
**The manufacturer has voluntarily withdrawn Grepa-
floxacin from the market because of potential risk of
cardiovascular events.
Methicillin-resistant
Staph
lococcus aureus
(MRSA)
V
ancomycin
Methicillin-resistant
Staph lococcus epidermidis
V
ancomycin
Enterococci
(Group D Streptococci)
1. Ampicillin
2. Vancomycin
there is now emerging
resistance to vancomycin
3. Imipenem and Meropenem
4. Piperacillin
5. Levofloxacin,
Trovafloxacin,*
Grepafloxacin,** and
Sparfloxacin
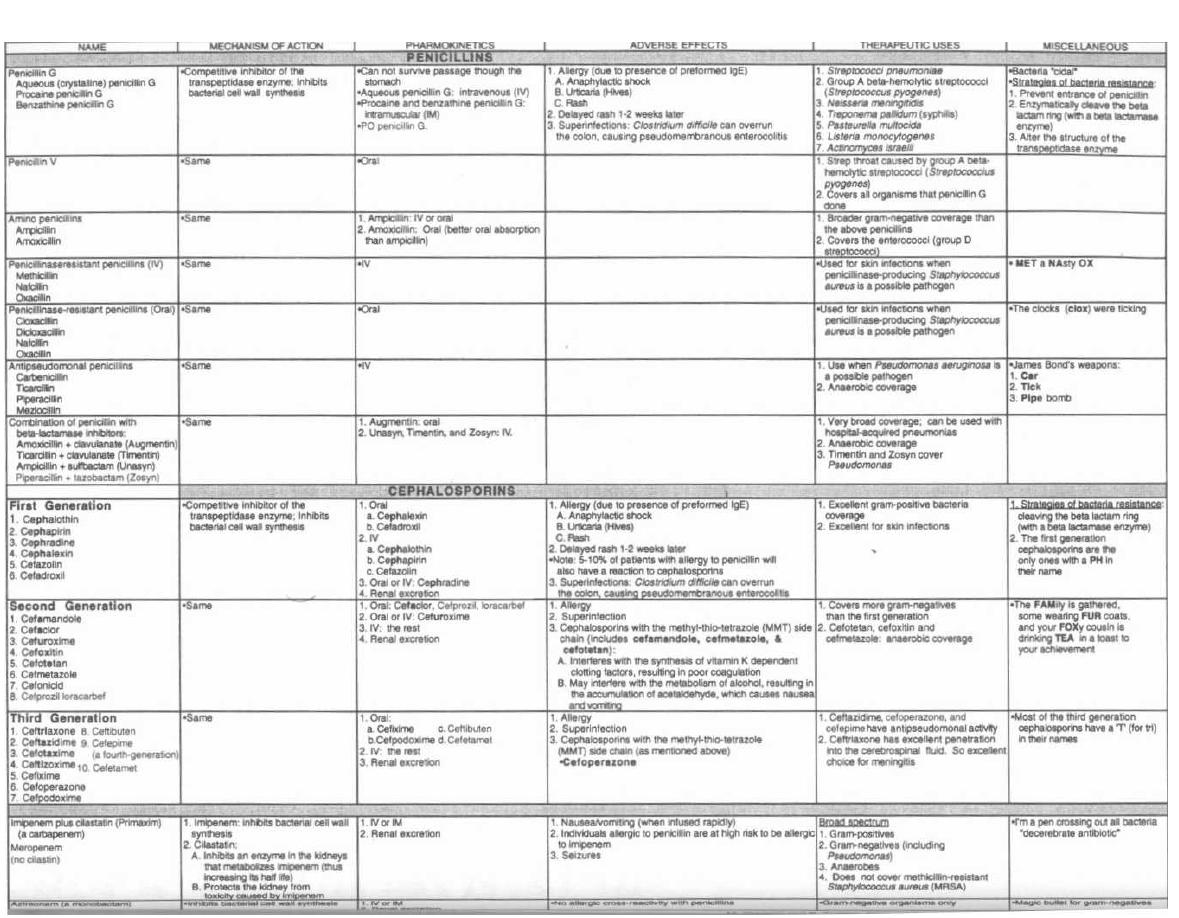
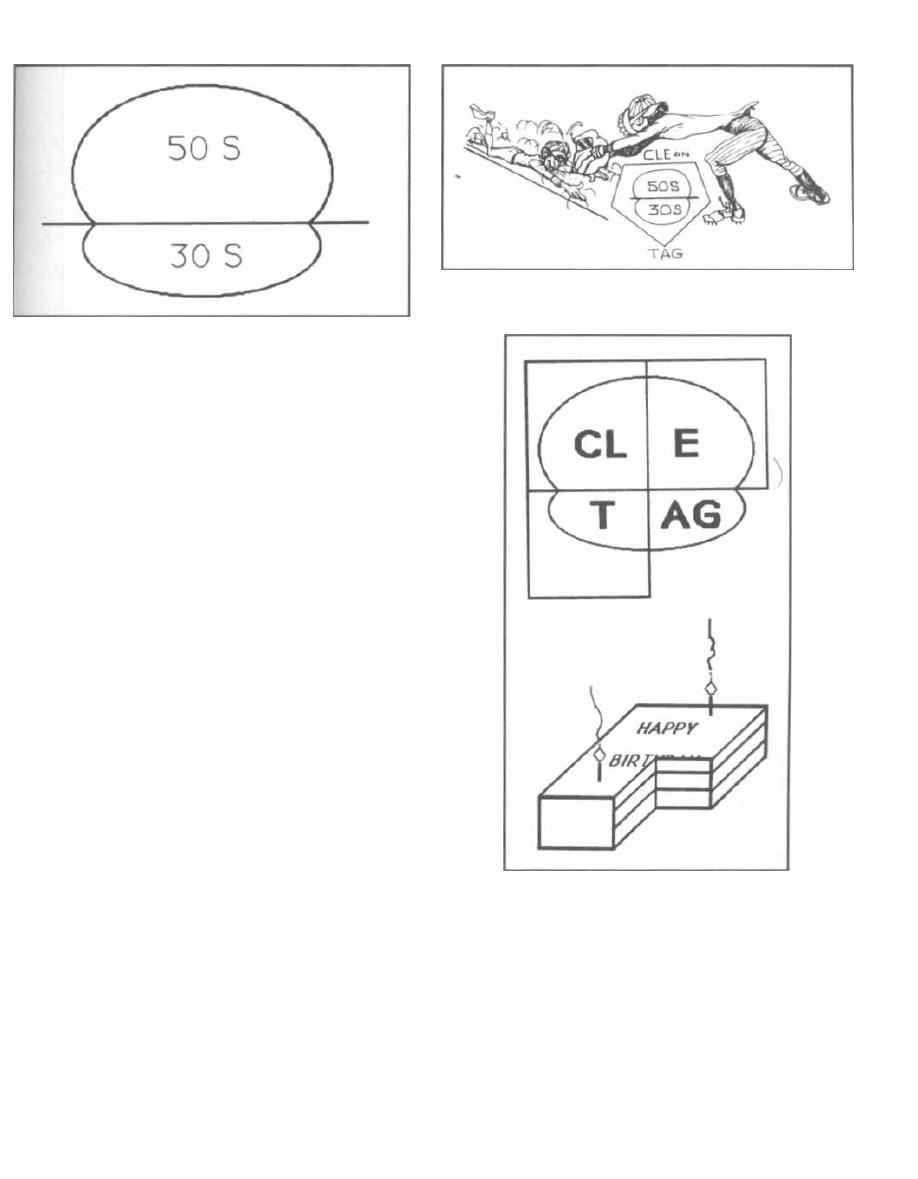
Figure 17-1
All cells depend on the continued production of proteins
for growth and survival. Translation of mRNA into the
polypeptides that make up these proteins requires the
use of ribosomes. Antibiotics that inhibit ribosomal ac-
tion would thus inhibit cellular growth and survival.
Since we only want to inhibit the growth of pathogenic
bacterial cells during an infection and not our own cells,
we are fortunate that bacteria actually have a different
type of ribosome than we do. We can exploit this differ-
ence by specifically inhibiting the ribosomes of bacteria,
while sparing the function of our own ribosomes. Bacte-
rial ribosomes are smaller than ours. While we have an
80S particle, the bacterial ribosome consists of a 70S
particle that has 2 subunits: the 50S (large) and the 30S
(small). (Surprisingly, 50S + 30S = 70S)
Fig. 17-1. The bacterial ribosome.
There are 5 important types of antibiotics that inhibit
the function of the bacterial ribosome. Three of them in-
hibit the large 50S subunit, and the other two inhibit
the small 30S subunit.
Here's how you can remember these 5 drugs:
Fig. 17-2. Convert the ribosome to home plate and
picture a baseball player sliding into home. The ball is
fielded by the catcher, who makes a CLEan TAG and
the player is out!!! Here is what CLEan TAG helps you
remember:
C for Chloramphenicol and Clindamycin
L for Linezolid
E for Erythromycin
T for Tetracycline
AG for Aminoglycosides
Note that the word CLEan lies over the base and the
word TAG beneath the base. This corresponds to the ri-
bosomal subunit that these drugs inhibit: CLEan in-
hibits the 50S; TAG inhibits the 30S.
CHAPTER 17. ANTI-RIBOSOMAL ANTIBIOTICS
125
Figure 17-2
Figure 17-3
Fig. 17-3. To remember which of these are orally ab-
sorbed, we have drawn boxes around the CLEan TAG on
the ribosome. Notice that the boxes do not extend
around the aminoglycosides (AG). We now draw a cake
with one-fourth missing-the same quadrant that is
missing above. You can eat three quarters of the cake.
The fourth piece (representing the aminoglycoside
quadrant) is missing, as this is the one anti-ribosomal

antibiotic that cannot be absorbed orally. The amino-
glycoside must be given IM or IV for systemic treatment
of infections.
Chloramphenicol
(The "Chlorine")
This drug has an amazing spectrum of activity. It is
one of the few drugs (like Imipenem) that kills most
clinically important bacteria. It is like pouring
"chlorine" on the organisms. Gram-positive, gram-neg-
ative, and even anaerobic bacteria are susceptible. It
is one of the handful of drugs that can kill the anaerobic
Bacteroides fragilis.
Clinical Uses
Because of its rare but severe side effects, this other-
wise excellent drug is used only when there is no alternate
antibiotic, and thus the benefits far outweigh the risks:
1) It is used to treat bacterial meningitis, when the
organism is not yet known and the patient has severe
allergies
to
the
penicillins,
including
the
cephalosporins. The wide spectrum of activity of chlo-
ramphenicol and excellent penetration into the CSF will
protect this patient from the devastating consequences
of meningitis.
2) Young children and pregnant women who have
Rocky Mountain spotted fever cannot be treated with
tetracycline due to the side effects of tetracycline dis-
cussed on page 128. Chloramphenicol then becomes the
drug of choice.
Note: In under-developed countries this drug is
widely used. It only costs pennies and covers every-
thing. Third world nations do not have the luxury of ex-
pensive alternative drugs available in the U.S.
Adverse Effects
Fig. 17-4. Picture a can of chloramphenicol chlorine.
Now picture the chlorine being poured down the shaft of a
long bone. You can well imagine that the bone marrow
would dissolve. This drug is famous for 2 types of bone
marrow depression. The first is dose-related and re-
versible, and often only causes an anemia. The second type
wipes out the bone marrow irreversibly and is usually fa-
tal. This is called aplastic anemia. Aplastic anemia
caused by chloramphenicol is extremely rare, occurring in
only 1:24,000 to 1:40,000 recipients of the drug.
Fig. 17-5.
Now picture a baby who leaps into a freshly
chlorinated pool. The baby crawls out of the pool, and
the chlorine has turned the baby's skin gray (Gray
Baby Syndrome). Neonates, especially preemies, are
unable to fully conjugate chloramphenicol in the liver or
excrete it through the kidney, resulting in very high
CHAPTER 17. ANTI-RIBOSOMAL ANTIBIOTICS
126
Figure 17-4
Figure 17-5

blood levels. Toxicity occurs with vasomotor collapse
(shock), abdominal distention, and cyanosis, which ap-
pears as an ashen gray color.
Clindamycin
Clinical Uses
This drug is NOT useful against gram-negative bugs.
So what is it good for? Many gram-positive bugs are in-
hibited. So what? What else?!!!?
Anaerobic infections! This is another of the rare
handful of antibiotics that cover anaerobes (including
Bacteroides fragilis).
Surgeons use clindamycin along
with an aminoglycoside for penetrating wound infec-
tions of the abdomen, which may occur with bullet and
knife trauma. When the GI tract is perforated, it re-
leases its contents of gram-negative and anaerobic bugs
into the sterile peritoneal cavity. The aminoglycosides
cover the aerobic gram-negative organisms, and clin-
damycin covers the anaerobes.
Clindamycin is also used for infections of the female
genital tract, such as septic abortions, as there are a lot
of anaerobes there. Oral preparations of clindamycin
and vaginal cream are alternatives to metronidazole
for the treatment of bacterial vaginosis. Topical clin-
damycin solution is also useful in the treatment of acne
vulgaris and rosacea (adult acne).
Adverse Effects
You must know this: Clindamycin can cause
Pseudomembranous Colitis!!!!!
When you give a patient clindamycin, or another po-
tent antibiotic for that matter, it will destroy the nat-
ural flora of the GI tract.
Clostridium difficile,
if
resistant to clindamycin, will grow like crazy and se-
crete its exotoxin in the colon. This exotoxin causes ep-
ithelial cell death and colonic ulcerations that are
covered with an exudative membrane; thus the name
pseudomembranous colitis. These patients often pre-
sent with a severe diarrhea. Stool cultures yielding
Clostridium dificile
or titers of toxin found in the stool
can help establish a diagnosis.
Note: While clindamycin was first identified as the
cause of pseudomembranous colitis, it is noteworthy
that other antibiotics also cause this condition. In fact
most cases are now caused by the penicillin fam-
ily drugs because they are prescribed more frequently.
To treat pseudomembranous colitis, you must give oral
vancomycin or metronidazole. Vancomycin passes through
the GI tract without being absorbed and is therefore
highly concentrated upon reaching the colon. The high
concentration can overwhelm and kill
Clostridium diffi-
cile. Metronidazole is less expensive and is now the
preferred agent, because use of oral vancomycin may con-
tribute to vancomycin resistant enterococcus!
CHAPTER 17. ANTI-RIBOSOMAL ANTIBIOTICS
12 7
Figure 17-6
Fig. 17-6.
Visualize a VAN (vancomycin) and a
METRO (metronidazole) cruising down the GI tract.
They run over the ulcerative potholes of pseudomembra-
nous colitis and kill the offending
Clostridium dificile.
Linezolid
("The Godzilla Lizard")
Clinical Uses
Linezolid, the Godzilla Lizard, is a newer antimicro-
bial agent for stamping out resistant gram positive
bugs. Linezolid blocks the 50S ribosomal subunit and
thus has activity against gram positive organisms in-
cluding those resistant to other antimicrobials. The
Lizard will likely find a place as a last resort for van-
comycin resistant enterococcus (VRE).
Adverse Effects
Headache occuring in 27% of patients and GI upset
(nausea, vomiting and diarrhea) in up to 18% of patients
are the most common side effects seen to date.
Erythromycin
("A Wreath")
Clinical Uses
Gram-positive organisms absorb erythromycin 100
times better than do gram-negative bugs. It is inactive
against most gram-negatives. You will use this drug of-
ten, for it covers gram-positive bacteria,
Mycoplasma,
and the gram-negatives
Legionella
and
Chlamydia,
also
known as atypicals.
Erythromycin is the drug of choice for community-ac-
quired pneumonia that does not require hospitalization.
This is because it covers
Streptococcus pneumoniae, My-
coplasma pneumoniae,
and
Chlamydia trachomatis
(strain TWAR), all common causes of community-ac-
quired pneumonia.
Erythromycin is often used as an alternative to peni-
cillin for streptococcal and staphylococcal organisms in

Figure 17-7
penicillin-allergic patients, especially for strep throat
and cellulitis.
Fig. 17-7. Erythromycin is the drug of choice for Le-
gionnaires' disease (see Chapter 10). The heroic
French foreign legionnaire has died in a desert battle.
In his honor, a wreath is laid by his grave. Notice the
tomb stone is in the shape of a cross to help you re-
member that erythromycin covers gram-positive organ-
isms (and don't forget atypicals! Tomb stone courtesy of
Dr. Cornejo, U. of Colorado).
Adverse Effects
Erythromycin is one of the safest antibiotics, a
pretty wreath compared to that nasty chlorine. Its few
side effects include:
1) Common and dose-dependent abdominal pain
( GI irritation) resulting from stimulation of intestinal
peristalsis.
2) Rare cholestatic hepatitis. Imagine a wreath slip-
ping into the bile duct and blocking flow.
There are now new drugs in this class (the macrolide
antibiotics) such as: clarithromycin, azithromycin,
and roxithromycin. They are showing promise in
treating the same bugs plus severe staphylococcal in-
fections,
H. influenzae,
and even coverage of some of the
atypical mycobacterium
( Mycobacterium avium-intra-
cellulare, MAI).
Azithromycin can be used as an alternative to doxycy-
cline for the treatment of (chlamydial) non-gonococcal ure-
thritis. It can be given as a single dose by mouth. It is also
used commonly to treat community acquired pneumonia.
CHAPTER 17. ANTI-RIBOSOMAL ANTIBIOTICS
12 8
Tetracycline/Doxycycline
("The Tet Offensive")
Tetracycline chelates with cations in milk and milk
products, aluminum hydroxide, Ca'+, and Mg *. When
it is chelated, it will pass through the intestine without
being absorbed. Doxycycline is a tetracycline that
chelates cations poorly and is thus better absorbed with
food. IV tetracycline is no longer available.
Clinical Uses of Doxycycline
This drug is used for all the diseases you would expect
a young soldier in the Tet offensive to get by crawling
around in the jungle and mingling with prostitutes on
leave:
1) Venereal diseases caused by
Chlamydia tra-
chomatis.
2) Walking pneumonia caused by
Mycoplasma pneu
-
moniae
(used as an alternative to erythromycin).
3) Animal and tick-borne diseases caused by
Bru-
cella
and
Rickettsia
(see the ticks on the soldier's pants
in Fig. 17-8).
4) Doxycycline also works wonders for acne.
Adverse Effects
Fig. 17-8. Picture a Vietcong soldier involved in the
Tet offensive to help remember these important side
effects:
1) This soldier is naturally very nervous as the Tet of
fensive involved waves of soldiers running into 20t-century
American fire power. So he has GI irritation with nausea,
vomiting, and diarrhea. This is a common side effect.
2) A grenade has blown up near him, burning his
skin like a sunburn. Notice the rays of light going from
the explosion to his face. Phototoxic dermatitis is a
skin inflammation on exposure to sunlight.
3) Shrapnel has struck his kidney and liver: renal
and hepatic toxicity. These adverse effects are rare
and usually occur in pregnant women receiving high
doses by the intravenous route.
4) Note the dark discolored teeth of the soldier.
This drug will chelate to the calcium in the teeth and
bones of babies and children under age 7, resulting in
brown teeth and depressed bone growth. Don't give
the drug to pregnant women or their baby's teeth will
look like those of the soldier.
Aminoglycosides
(A
Mean Guy)
Aminoglycosides must diffuse across the cell wall to en
ter the bacterial cell, so they are often used with peni-
cillin, which breaks down this wall to facilitate diffusion.

Figure 17-8
Clinical Uses
In general, aminoglycosides kill aerobic gram-nega-.
tive enteric organisms (the enterics are the bugs that
call the GI tract home, such as E. coli and company).
The aminoglycosides are among the handful of drugs
that kill the terrible Pseudomonas aeruginosa!!!
Most aminoglycosides end with - mycin:
1) Streptomycin is the oldest one in the family.
Many bugs are resistant to it.
2) Gentamicin is the most commonly used of all the
aminoglycosides. It is combined with penicillins to treat
i n-hospital infections. There are also many bacterial
strains resistant to this drug.
3) Tobramycin
is
good
against
the
terrible
Pseudomonas aeruginosa.
4) Amikacin does not end with mycin (sorry).
Maybe that is to set it apart. It has the broadest spec-
CHAPTER 17. ANTI-RIBOSOMAL ANTIBIOTICS
129
trum and is good for hospital-acquired (nosocomial) in-
fections that have developed resistance to other drugs
while doing time in the hospital.
5) Neomycin has very broad coverage but is too
toxic, so it can only be used topically for:
a) Skin infections.
6) Netilmicin
b) Preoperative coverage before GI surgery. This
drug is given orally before GI surgery as it cruises
down the GI tract, without being absorbed, killing
the local inhabitants. This prevents spilling of or-
ganisms during surgery into the sterile peritoneal
cavity.
Adverse Effects
Here's how we will remember the side effects: Picture
this huge boxer, a mean guy ( Aminoglycoside), and
now check out these pictures:
Figure 17-9
Fig. 17-9.
In the eighth round A MEAN GUY deliv-
ers a crushing left hook to his opponent's ear, hurling
him off balance, ears ringing and head spinning (eighth
cranial nerve toxicity: vertigo, hearing loss). The hear-
i ng loss is usually irreversible.
Fig. 17-10.
With his opponent off balance , A mean guy
surges upward with a savage right hook into his left side,
pulverizing his kidney (renal toxicity). Aminoglycosides
are renally cleared and can damage the kidney. This can
be reversible, so always follow a patient's BUN and crea-
tinine levels, which increase with kidney damage.
Fig. 17-11.
The opponent drops to the floor, out cold
i n a complete neuromuscular blockade, unable to
move a muscle, or even breathe. This curare-like effect
is rare.
Note: These side effects occur if the dose is very high,
so when using these in the hospital, the drug level in the
blood is checked after steady state levels have been
achieved (usually after the third dose). With appropri-
ate blood levels, these agents are generally safe.
CHAPTER 17. ANTIRIBOSOMAL ANTIBIOTICS
13 0
Figure 17-10
Figure 17-11
Spectinomycin
(Spectacular Spectinomycin)
This drug has a name that sounds like an aminogly-
coside, but it is different structurally and biologically.
Its mechanism is similar in that it acts on the 30S ribo-
some to inhibit protein synthesis, but exactly how is not
known. Group this with the aminoglycosides in the
CLEan TAG mnemonic (see Fig. 17-2) to remember its
action, but note that it is NOT an aminoglycoside. It is
given as an IM injection.

Figure 17-12
Clinical Uses
Spectinomycin is used to treat gonorrhea, caused by
Neisseria gonorrhoeae, as an alternative to penicillin
and tetracycline (doxycycline), since many strains are
resistant to these drugs.
Fig. 17-12.
Mr. Gonorrhoeae, resistant to tetracycline
and penicillin.
Fig. 17.13.
Spectacular spectinomycin treats resis-
tant Neisseria gonorrhoeae.
Let's briefly review the treatment of gonococcal ure-
thritis (gonorrhea) since this will incorporate a lot of the
drugs we have studied.
A patient presents with burning on urination and a
purulent penile discharge. When you Gram stain the
Figure 17-13
CHAPTER 17. ANTI-RIBOSOMAL ANTIBIOTICS
13 1
discharge, you see tiny red (gram-negative) kidney-
shaped diplococci inside the white blood cells. Now
what? There are many penicillinase-producing and
tetracycline-resistant Neisseria gonorrhoeae, but you
still have a few antibiotics to chose from:
1) Ceftriaxone (a third generation cephalosporin):
Give one shot IM in the butt! Also give doxycycline by
mouth for 7 days to get the Chlamydia trachomatis that
is hiding in the background in 50% of cases of urethritis!
Azithromycin can be used as an alternative to doxycy-
cline. It can be given as a single dose by mouth. Or:
2) Quinolone antibiotics (ciprofloxacin, ofloxacin)
get Neisseria gonorrhoeae and are given as one oral dose
(along with doxycycline for the Chlamydia). Or:
3) Spectinomycin: Give one shot in the butt! (along
with doxycycline for the Chlamydia).
Adverse Effects
Infrequent and minor. Spectinomycin does NOT
cause the vestibular, cochlear, and renal toxicity that
the aminoglycosides do.
Fig. 17-14.
Summary of anti-ribosomal antibiotics.
Recommended Review Articles:
Alvarez-Elcoro S, Enzler MJ. The macrolides: Erythromycin,
Clarithromycin, and Azithromycin. Symposium on antimi-
crobial agents. Mayo Clin Proc
1999;74:613-634.
Edson RS, Terrell CL. The aminoglycosides. Symposium on
antimicrobial agents. Mayo Clin Proc
1999;74:519-528.
Kasten MJ. Clindamycin, Metronidazole, and Chlorampheni-
col. Symposium on antimicrobial agents. Mayo Clin Proc
1999;74:825-833.
Smilak, JD. The tetracyclines. Symposium on antimicrobial
agents. Mayo Clin Proc
1999;74:727-729.
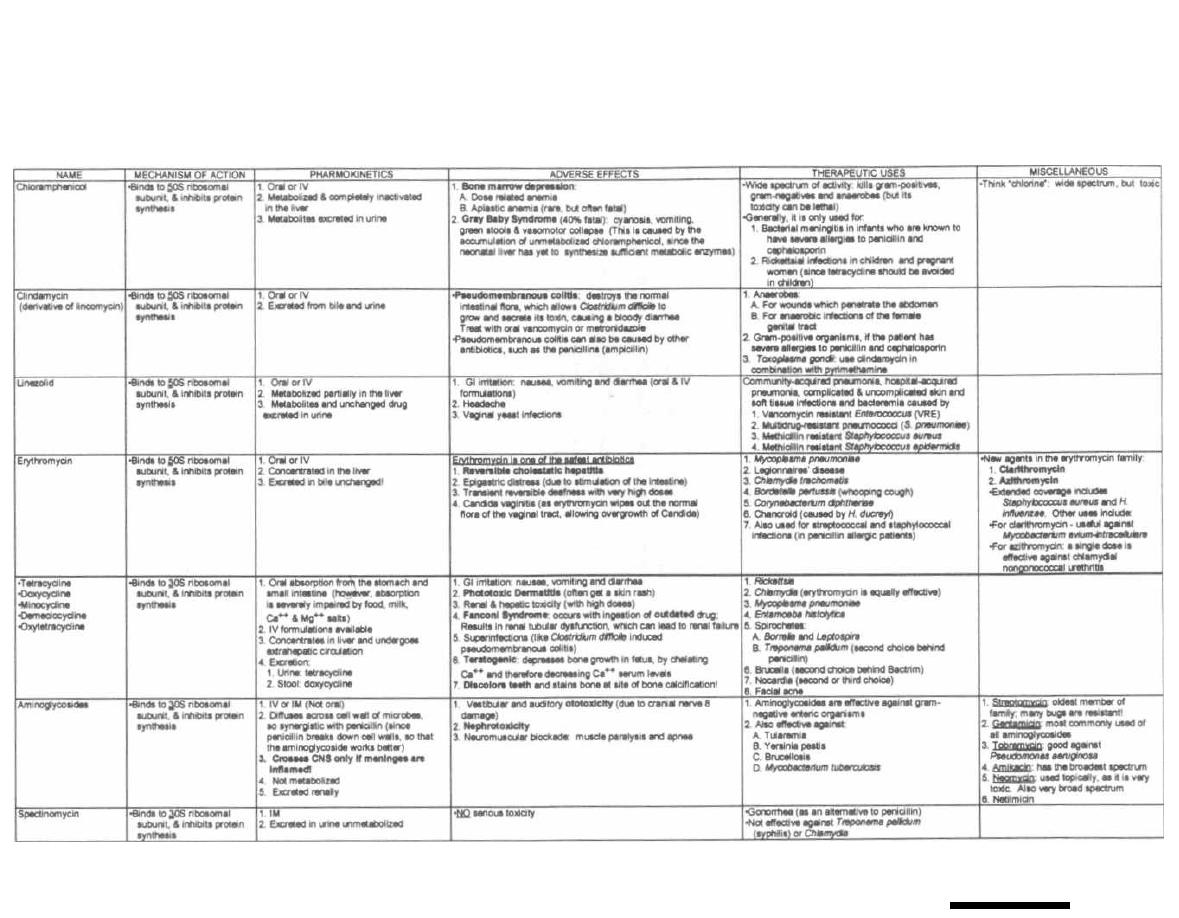
Figure 17-14 ANTI-RIBOSOMAL DRUGS
M. Gladwin and B. Trattler, Clinical
Microbiology Made Ridiculously
Simple ©MedMaster

CHAPTER 18. ANTI-TB and ANTI-LEPROSY ANTIBIOTICS
Figure 18-1
TREATMENT OF TUBERCULOSIS
This chapter will cover the first-line anti-tuberculosis
antibiotics and the logical approach to their use.
The first-line drugs, in order of their frequency of use,
are:
Isoniazid (INH)
"I saw a
Rifampin
Red
Pyrazinamide
Pyre-BURNING THE
LIVER"
Ethambutol
Streptomycin
Fig. 18-1. Isoniazid ("I saw"), Rifampin ("red"), and
Pyrazinamide ("pyre"), are first-line anti-tuberculosis
antibiotics that can cause liver damage ("burning the
liver").
When it comes to tuberculosis, you will encounter 2
populations of patients: 1) those with active tuberculo-
sis and 2) those with a reactive PPD skin test, repre-
senting a latent infection. These 2 populations are
treated very differently.
Treatment of Active Tuberculosis
A patient presents with dyspnea, fever, productive
cough, and night sweats that have lasted 2 months,
along with upper lobe consolidation on chest X-ray.
Acid-fast bacilli are identified from a sputum sample.
A patient with active pulmonary or extra-pulmonary
tuberculosis should receive a 6-month or 9-month treat-
ment as follows:
6-month regimen: 2 months of isoniazid, rifampin,
and pyrazinamide, followed by 4 months of isoniazid
and rifampin.
9-month regimen: 9 months of isoniazid and
rifampin.
Notice that the 2 regimens differ in the inclusion or
exclusion of pyrazinamide. Pyrazinamide is rapidly bac-
133
teriocidal to Mycobacterium tuberculosis, but the risk of
liver toxicity is too great if used for more than 2 months.
Treatment of PPD Reactors
These persons may have latent Mycobacterium tuber-
culosis
in their bodies and might develop a reactivation
tuberculosis. Treatment of PPD reactors is th us pre-
ventive. Isoniazid is usually used alone for 6-12
months as prophylactic therapy. Recently, a study
showed that a 2 month course of rifampin plus pyrazi-
namide was as effective as a 12 month course of isoniazid
in PPD positive patients. This is important due to the
high rate of non-compliance with a 12 month treatment
regimen.
Here is the difficulty: Not all persons who react to the
PPD test should be treated. Some of these persons will
never develop reactivation tuberculosis and the drugs
carry risks!!!
The decision to treat PPD reactors involves balancing
the risk of developing isoniazid-induced liver injury
against the risk of developing reactivation tuber-
culosis. Imagine a set of scales. On one side weighs the
)risk of developing isoniazid-induced hepatitis and on the
other side the risk of reactivating the disease tuberculosis.
Risk of Isoniazid Hepatitis
As indicated below, advancing age and alcohol con-
sumption increase the risk of developing hepatitis from
isoniazid and tip the scales towards not treating. No-
tice that under the age of 35 there is virtually no risk of
developing hepatitis with isoniazid:
AGE
% that develop HEPATITIS
<20
Rare
20-34
<0.3%
35-49
< 1.2%
>50
<2.3%

CHAPTER 18.
ANTI-TB AND ANTI-LEPROSY ANTIBIOTICS
Data from the Tuberculosis Advisory Committee re-
port (1974).
Risk of Reactivation Tuberculosis
Factors that increase the risk of reactivation include
recent PPD conversion, having fibrotic scars on chest
X-ray, exposure to household members with active tu-
berculosis, and being immunosuppressed. The larger
the skin reaction to PPD the more likely the test
is a true positive, representing
M. tuberculosis
infection. Because. these factors increase the risk of re-
activation of tuberculosis, they will tip the scales to-
wards treatment.
The Center for Disease Control and American
Thoracic Society have used these concepts to formulate
the following recommendations for treating patients
with a 6-12 month course of prophylactic isoniazid:
GREATEST RISK OF REACTIVATION: Treat the fol-
lowing patients at any age if PPD >_ 5mm:
1) Persons with HIV infection.
2) Persons with fibrotic changes on chest X-ray com-
patible with old healed tuberculosis.
3) Close contacts of persons with newly diagnosed ac-
tive tuberculosis. Note: In this case (especially with chil-
dren), even if the PPD is negative, treat for 3 months,
then repeat the PPD. If at that time it is < 5mm, the
isoniazid may be discontinued.
MODERATE RISK: Treat these patients at any age if
PPD 10mm:
1) Persons with medical conditions that lower
the immune system, like diabetes, prolonged steroid
or immunosuppressive treatment, renal failure, and
others.
2) Persons with recent skin test conversion within
the last 2 years.
3) Persons who inject drugs.
LESS RISK: Treat these patients if age < 35 and
PPD 10mm:
High-risk populations such as the homeless, resi-
dents of long-term care facilities, such as prisons and
nursing homes, and foreign-born from countries with a
high-prevalence of tuberculosis.
LOWEST RISK: Treat at the physician's discretion if
age < 35:
A person with no known risk factors and a PPD >
15mm.
134
Isoniazid-Resistant Organisms
Resistance to INH and the other antibiotics is devel-
oping. This should be suspected in persons from Africa,
Asia, or South America; homeless persons and others ex-
posed to resistant organisms; and those whose sputum
cultures remain positive after 2 months of treatment.
1) Culture susceptibility testing should follow the
initiation of treatment.
2) If resistance is suspected, 4 or more first-line
drugs should be used (INH, rifampin, pyrazinamide,
ethambutol, or streptomycin).
3) If resistance develops, never add a single new an-
tibiotic; always add two. This will insure that the re-
sistant
M. tuberculosis will
be unable to develop further
resistance.
Note that there are now multiply resistant
M. tu-
berculosis
organisms that may require 4-5 different
antibiotics.
DOT the I's and Cross the T's to Prevent
Resistance!!!
DOT or Directly Observed Therapy: Health care
providers in outpatient settings have their patients
come into the clinic to receive their medications under
direct observation to ensure adherence. Numerous
studies have now documented that this strategy can
decrease resistance and increase treatment efficacy.
ANTI-TB ANTIBIOTICS
Isoniazid, Rifampin, and Pyrazinamide:
1) All cause hepatotoxicity: Patients on these
medications must understand the symptoms of hepati-
tis so they can report to a doctor immediately should
they develop. Mild elevations of liver enzymes can be ex-
pected to occur in 15-20% of patients on isoniazid, but
should these levels exceed 3-5x the upper limit of nor-
mal, the drugs should be discontinued.
2) All are absorbed orally: This is very important
since these must be administered for 6-9 months. They
must be orally absorbed!!!
3) All penetrate into most tissues: They must
reach the center of caseous granulomas.
Isoniazid (INH)
("I Saw")
This is a great antibiotic because it is inexpensive, ab-
sorbed orally, and bacteriocidal. Were it not for the tox-
icity, it would be perfect!!!
INH interferes with the biosynthesis of the mycolic
acid component of the cell wall of the Mycobacteria.

Adverse Effects
1) What do you think? Hepatotoxicity!!! Alcoholics
beware! Alcohol increases the metabolism of INH by the
liver, which increases the risk of developing hepatitis
and decreases the therapeutic effect.
2) INH increases the urinary excretion and depletion
of pyridoxine (vitamin B6), which is needed for proper
nerve function. INH will thus lead to decreased B6 lev-
els, characterized by peripheral neuropathy, rash,
and anemia. Many Docs routinely give Bs vitamins
with INH.
Rifampin
!`Red")
Think Rifampin:
1) Red: Body fluids such as urine, feces, saliva,
sweat, and tears are colored a bright red-orange color by
rifampin. This is not harmful to the patient, but pa-
tients must be made aware of this or they will discon-
tinue the medicine in a panic.
2) RNA: Rifampin inhibits the DNA-dependent RNA
polymerase of the Mycobacterium tuberculosis bugs.
Adverse Effects
1) Hepatitis (much less than INH).
2) Rifampin induces the cytochrome P450 enzyme
system (also called the microsomal oxidase system, or
MOS), so many other drugs are gobbled up by the
spruced-up MOS. This results in decreased half-lives
of certain drugs in patients taking rifampin. Some
examples:
a) Coumadin (an anticoagulant): Blood-thinning
effect will be reduced.
b) Oral contraceptives: Women can get pregnant
and get breakthrough bleeding!
c) Oral hypoglycemics and corticosteroids are less
effective.
d) Anticonvulsants such as phenytoin (seizures!).
Rifabutin
Rifabutin is very similar to rifampin in structure, an-
tibacterial activity, metabolism, and adverse reactions.
Itis commonly used in the treatment of Mycobacterium
auium
•i
ntracellulare
( MAI). MAI is usually more sensi-
tive to rifabutin than rifampin.
The same drug-drug interactions of rifampin
should be considered for rifabutin however, rifabutin
induces cytochrome P450 less than rifampin. Thus
rifabutin is helpful in treating patients with tubercu-
losis and HIV.
CHAPTER 18.
ANTI-TB AND ANTI-LEPROSY ANTIBIOTICS
135
Figure 18-2
Pyrazinamide
("Pyre")
The mechanism of action of pyrazinamide is not
known.
Adverse Effects
Pyrazinamide is hepatotoxic (no kidding?!) This med-
icine is usually given for no more than 2 months to avoid
liver toxicity. Avoid it in pregnancy (unknown effect on
fetus).
Ethambutol
("Ethane-Butane Torch")
Adverse Effects
Fig. 18-2.
The main side effect of ethambutol is a
dose-dependent, reversible, ocular toxicity. Think of an
ethane-butane flame torch, torching an eye. The ocu-
lar toxicity is manifested by:
1) Decreased visual acuity with loss of central vision
(central scotomata).
2) Color vision loss.
Ethambutol is not used in young children because
they are not able to report vision deterioration. Adults
are tested for visual acuity and color perception
at regular intervals. Many doctors instruct their pa-
tients to read the fine newspaper print everyday as a
self exam.
Streptomycin
Streptomycin is in the aminoglycoside family, which
inhibits protein synthesis at the 30S ribosomal subunit,
and is given IM or IV. It is ototoxic and nephrotoxic (see
Chapter 17). Avoid it in pregnant women (can cause
congenital deafness).

Fixed-Dose Combinations
Fixed-dose combinations are available as Rifamate
(isoniazid and rifampin) and Rifater (isoniazid, rifam-
pin, and pyrazinamide). Such combinations are strongly
encouraged for adults who are self-administering their
medications because they may enhance adherence, re-
duce the risk of inappropriate monotherapy, and pre-
vent drug resistance.
Second-line Drugs
These can be used when multiple antibiotics are
needed for the treatment of multi-drug resistant My-
cobacterium. tuberculosis.
Para-aminosalicyclic acid
Capreomycin sulfate
Cycloserine
Ethionamide
Kanamycin
Amikacin (aminoglycoside)
Quinolones such as ciprofloxacin and ofloxacin
Treatment of LEPROSY
Three drugs are used in the treatment of leprosy: dap-
sone, rifampin, and clofazimine.
The Rap Zone of Dapsone
Where's your ears?
There on the floor.
Where's your hand?
I left it on the door.
Come on Doe, look and see,
these rappin' clowns got leprosy.
Hey, you clown!
You left your nose in the car,
Hey, you clown!
You left your toes in a bar,
Call the doc, to the Rap Zone,
Your first-line drug remains dapsone.
And if you dance to this stupid rappin',
watch your feet as they start a stampin',
There's only one thing to help your dancin,
Time to reach for the drug, rifampin.
Their peelin' clown faces look really lean.
They're healing faster than can be seen,
As long as they stay close to clofazimine.
Severe cases of leprosy should be treated with ri-
fampin, dapsone, and clofazimine for a minimum of 2
years and until patients are acid-fast bacilli negative.
CHAPTER 18. ANTI-TB AND ANTI-LEPROSY ANTIBIOTICS
13 6
Figure 18-3
Less severe cases are treated with rifampin and dap.
sone for 6 months.
See anti-tuberculosis medications (page 134), for
more on rifampin. See the sulfa drugs (Chapter 19) for
more on dapsone.
Clofazimine
Fig. 18-3.
A clown-faced clown climbs a DNA double
helix stairway. His outfit is colored red and black:
1) Clofazimine works by binding to the DNA of My
cobacterium Leprae. It also has anti-inflammatory
actions that are helpful in treating the
leprosy
reactions.
2) Clofazimine is a red-colored compound, and when
it deposits in the skin and conjunctiva, it colors these
tissues red. Any place on the body where there is a lep-
rosy lesion, the skin will appear tan to black. Note the
clown's red and black outfit.

CHAPTER 18. ANTI-TB AND ANTI-LEPROSY ANTIBIOTICS
Leprosy Reactions
Fifty percent of patients treated for leprosy develop a
leprosy reaction. There are 2 types (1 and 2) and both
are immune-mediated, possibly in response to the in-
crease in dead organisms with treatment. The reactions
i nvolve inflammation of the nerves, testicles, eyes,
joints, and skin (erythematous nodules).
Type 1 reactions occur only in borderline patients
( BT, BB, BL), and almost always occur during the first
year of treatment. The skin lesions of leprosy typically
swell, becoming more edematous, and occasionally ul-
cerate. Neuritis can also occur, leading to sensory or mo-
tor nerve loss. The type 1 reaction is thought to be a
delayed hypersensitivity reaction to the dead bacilli.
When this reaction occurs, patients can be treated with
prednisone or clofazimine. It is important that you do
NOT withdraw the anti-leprosy drugs if a leprosy reac-
tion occurs.
Type 2 reaction (called Erythema Nodosum Lep-
rosum) is associated with borderline lepromatous (BL)
and lepromatous leprosy (LL). Commonly, a painful
nodular rash erupts in a previously normal-appearing
area of skin, along with a high fever. Neuritis, orchitis,
arthritis, iritis, and lymphadenopathy can occur as well.
The type 2 reaction is thought to be an immune com-
plex-mediated reaction involving the deposition of the
i mmune complexes in tissues followed by complement
activation. These patients can also be treated with pred-
nisone or clofazimine. However, the treatment of choice
is thalidomide. This is the only use of thalidomide that
is condoned in the U.S. because it is a potent teratogen.
Again, the anti-leprosy antibiotics are NOT to be with-
drawn!
Fig. 18-4.
Summary of antibiotics for Mycobacteria.
References
Hopewell PC, Bloom BR. Tuberculosis and other Mycobacter-
ial Diseases. In: Murray JF, Nadel JA, eds. Textbook of Res-
piratory Medicine. 2nd ed. Philadelphia: W.B. Saunders Co.
1994:1094-1160.
Isoniazid-associated hepatitis: summary of the report of Tu-
berculosis Advisory Committee and special consultants to
the director, Centers for Disease Control. MMWR 23:97-98,
1974.
U.S. Department of Health and Human Services, Division of
Tuberculosis Elimination, Centers for Disease Control and
Prevention, American Thoracic Society. Core Curriculum
on Tuberculosis: What the clinician should know. 3rd Edi-
tion. Atlanta, Georgia, 1994.
Van Scoy, Robert, M.D., Wilkowske, Conrad, M.D.; Antituber-
culous Agents; Mayo Clin Proc 67:179-187, 1992.
Recommended Review Article:
Van Scoy RE, Wilkowske CJ. Antimycobacterial therapy.
Symposium on antimicrobial agents. Mayo Clin Proc 1999;
74:1038-1048.
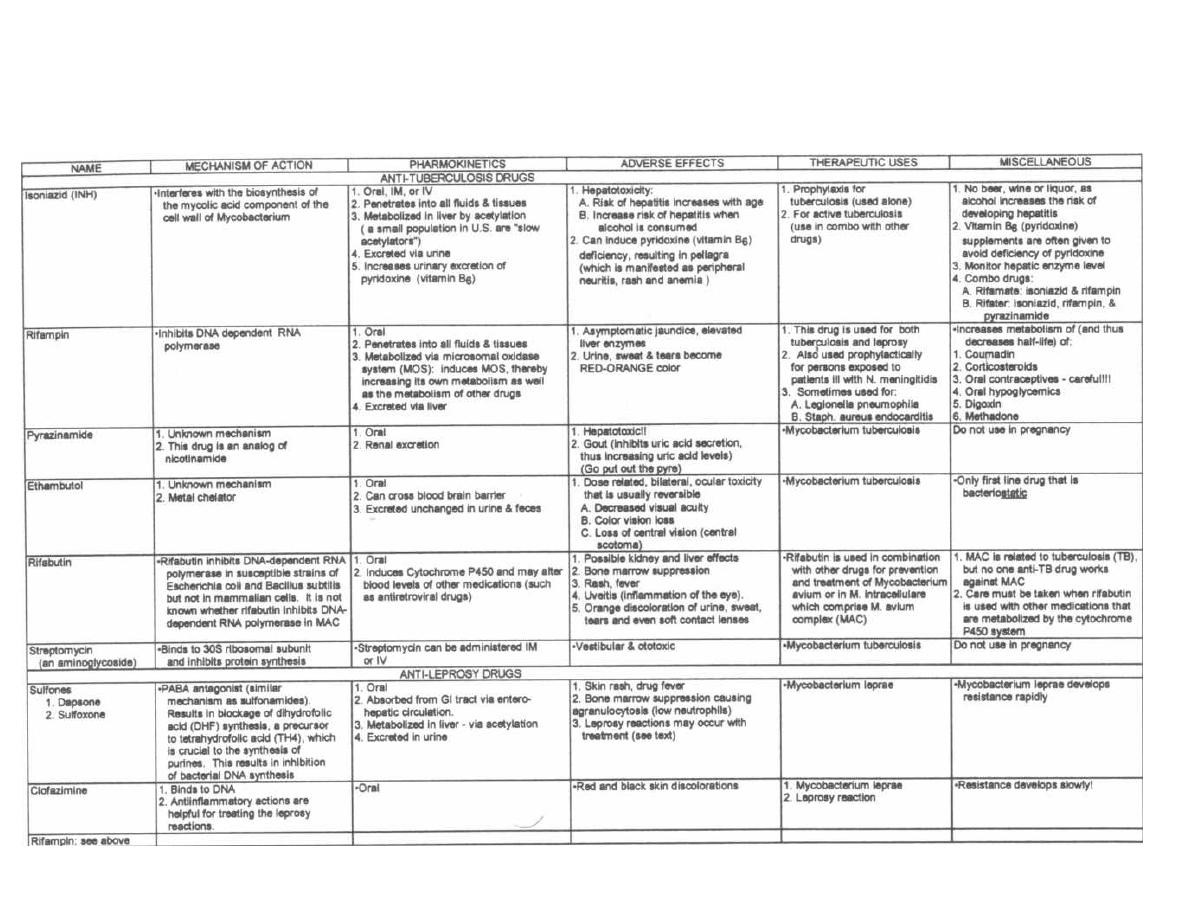
Figure 18-4 ANTIBIOTICS FOR MYCOBACTERIUM
M. Gladwin and B. Trattler,
Clinical
Microbiology
Made Ridiculously Simple
©MedMaster

CHAPTER 19.
MISCELLANEOUS ANTIBIOTICS
Figure 19-1
THE FLUOROQUINOLONE
ANTIBIOTICS
Ciprofloxacin and Family
This relatively new group of antibiotics is expanding.
The fluoroquinolones has become as large and impor-
tant a group as the penicillins and cephalosporins. The
reason for this is that they are safe, achieve high blood
levels with oral absorption, and penetrate extremely
well into tissues.
Fig. 19-1. All the antibiotics in the fluoroquinolone
family have the common ending -FLOXACIN. To help
you remember some facts about this drug family, think
of a crazy naked group of partyers, a FLOCK OF SIN-
NERS. They are all gyrating their hips as they party
and dance. The fluoroquinolones act by inhibiting DNA
gyrase, resulting in the breakage of the bacterial DNA
structure.
In the basic quinolone structure, the main feature
that distinguishes the fluoroquinolones from their
predecessor, nalidixic acid, is the addition of a fluo-
rine group, hence the name fluoroquinolone. The
139
evolution of quinolone structures now allow their
classification into first, second, third and fourth gen-
erations, with nalidixic acid solely in the first gener-
ation. Classification by generation is available on
page 143.
Resistance to the Fluoroquinolones
Like all antibiotics, with this excessive use, resis-
tant organisms are rapidly spreading. What makes
this particularly disconcerting is that the resistance
is against all the fluoroquinolones. Resistance is
caused by a point mutation in the bacterial DNA gy-
rase subunits. This powerful new family of antibi-
otics should be used carefully to reduce the spread of
resistance.
Adverse Effects
There are very few:
1) Some patients experience GI irritability (nausea,
vomiting, belly pain, diarrhea), as occurs with ery-
thromycin and doxycycline. The flock of sinners often
vomit after their excessive drinking.
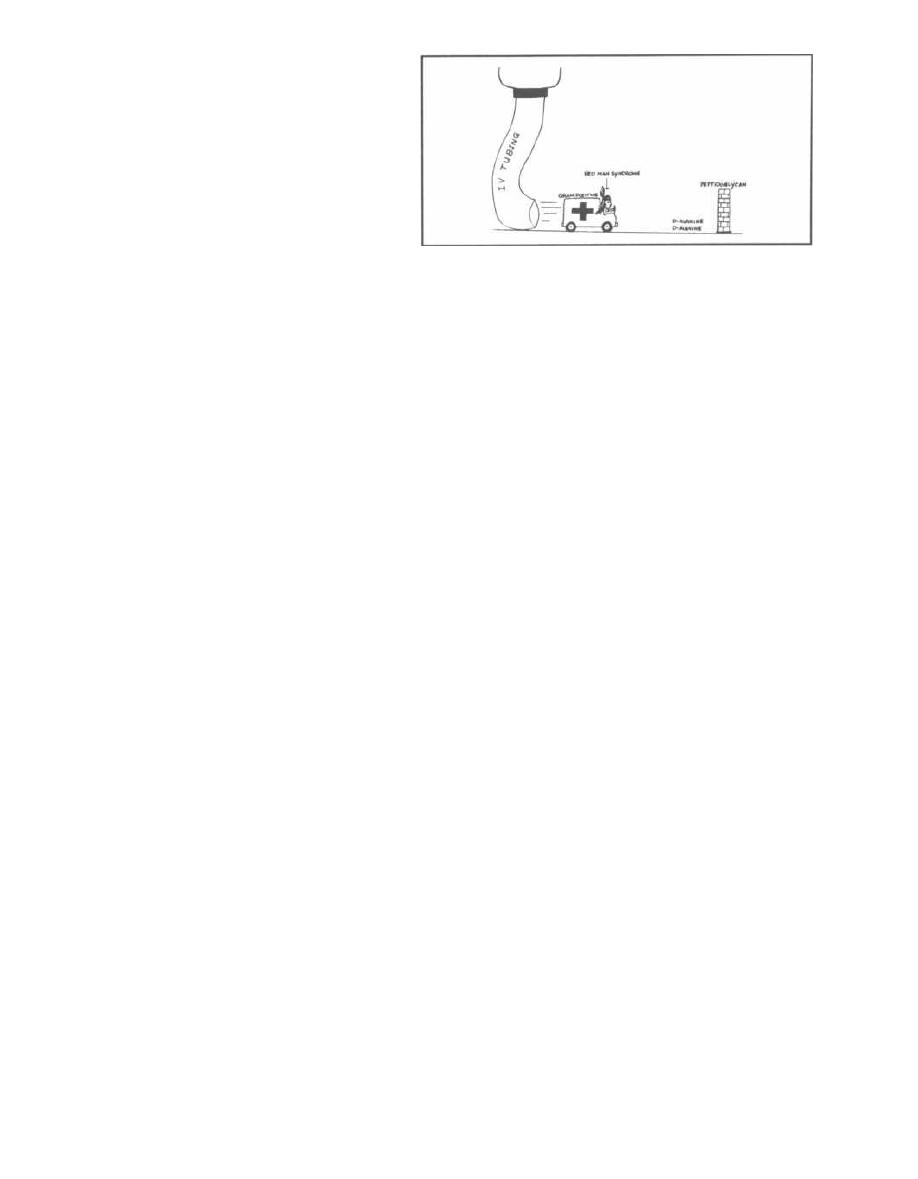
such as
Staphylococcus aureus,
gram-negatives including
Pseudomonas aeruginosa
(see Figure 16-15), and even
anaerobes such as
Bacteroides fragilis.
Add the treasure-
trove to your list of GORILLA-CILLINS!!! Unfortunately
the pirate who buried the treasure left behind a booby
trap that exploded when we opened the trove!
*Because of liver toxicity, the FDA advised that
trovafloxacin should be reserved for treatment ONLY in
patients that meet ALL of the following criteria:
Who have at least one of five types of serious and
life threatening infections listed below that is judged
by the treating physician to be serious and life or
li mb-threatening:
Nosocomial pneumonia (pneumonia acquired in the
hospital)
•
Community acquired pneumonia
•
Complicated intra-abdominal infections, including
post-surgical infections
Gynecological and pelvic infections
Complicated skin and skin structure infections, in-
cluding diabetic foot infections
**The manufacturer has voluntarily withdrawn
Grepafloxacin from the market because of potential
risk of cardiovascular events.
Sparfloxacin has two important side effects that set
it apart:
1) Up to 8% of patients develop mild to severe
photosensitivity.
2) It can prolong the Q-T interval on the EKG,
which can predispose a patient to the arrhythmia Tor-
sades de pointer.
1
VANCOMYCIN
This IV antibiotic has a critical role in the treatment
of infectious diseases. It is the opposite of aztreonam,
which covers all gram-negative bugs. Vancomycin
covers ALL GRAM-POSITIVE bugs!!!
Even MRSA (methicillin resistant
Staphylococcus
aureus)!!
Even the Enterococcus
( Streptococcus faecalis)!!
Even multi-resistant
Staphylococcus epidermidis
(in
infections of indwelling intravenous catheters).
It is also used to treat endocarditis caused by
Strepto-
coccus and
Staphylococcus
in penicillin-allergic patients.
Fig. 19-3.
A VAN with a + on its side (an ambulance
van) is driving out of some IV tubing. It is about to run
over an ear and hit a peptidoglycan cell wall. The
VAN is being driven by an Indian, the red man. This
picture helps us remember that VANCOMYCIN is given
N, kills gram-positive bugs by inhibiting peptidoglycan
production, and cause the red man syndrome. In the
CHAPTER 19. MISCELLANEOUS ANTIBIOTICS
14 1
Figure 19-3
latter, which follows rapid infusion of vancomycin , there
is often a nonimmunologic release of histamine, result-
ing in a red rash of the torso and itching skin. Slow in-
fusion over an hour or antihistamine premedication can
prevent this problem.
Vancomycin inhibits the biosynthesis of the gram-
positive peptidoglycan at a step earlier than penicillin.
It complexes with D-alanine D-alanine to inhibit
transpeptidation. Like penicillin, it acts synergisti-
cally with the aminoglycosides.
Vancomycin is not absorbed orally. We take ad-
vantage of this in the treatment of
Clostridium dificile
pseudomembranous colitis. Vancomycin is taken orally,
cruises down the GI tract unabsorbed, and kills the
Clostridium dificile!!
With the extensive use of vancomycin, new strains
of multiple drug-resistant gram-positive organisms
have emerged (see page 27). Synercid (quinopristin/
dalfopristin) is a new type of antibiotic that has a wide
spectrum of activity. It appears to be effective against
most of the multiple drug-resistant strains.
Linezolid is another new type of antibiotic in a to-
tally new class of agents with activity against gram-
positive organisms, including those resistant to other
antimicrobials (see Chapter 17).
ANTIMETABOLITES
Trimethoprim and Sulfamethoxazole
Nucleotide and DNA formation require tetrahydrofo-
late (TH4). Bacteria make their own TH4 and use Para
Amino Benzoic Acid (PABA-you know, the stuff in sun-
screen) to make part of the TH4. People don't make
TH4; we get it as a vitamin in our diet.
Fig. 19-4.
The Sulfa drugs look like PABA.
When you give a person one of the sulfa drugs (e.g., sul-
famethoxazole), the bacteria use it, thinking it's PABA.
( There isn't much room for higher cortical neurons in a
single-celled creature.) The sulfa drug competitively in-
hibits production of TH4. Since our cells don't make TH4,
it doesn't affect us, but it affects the bacteria.
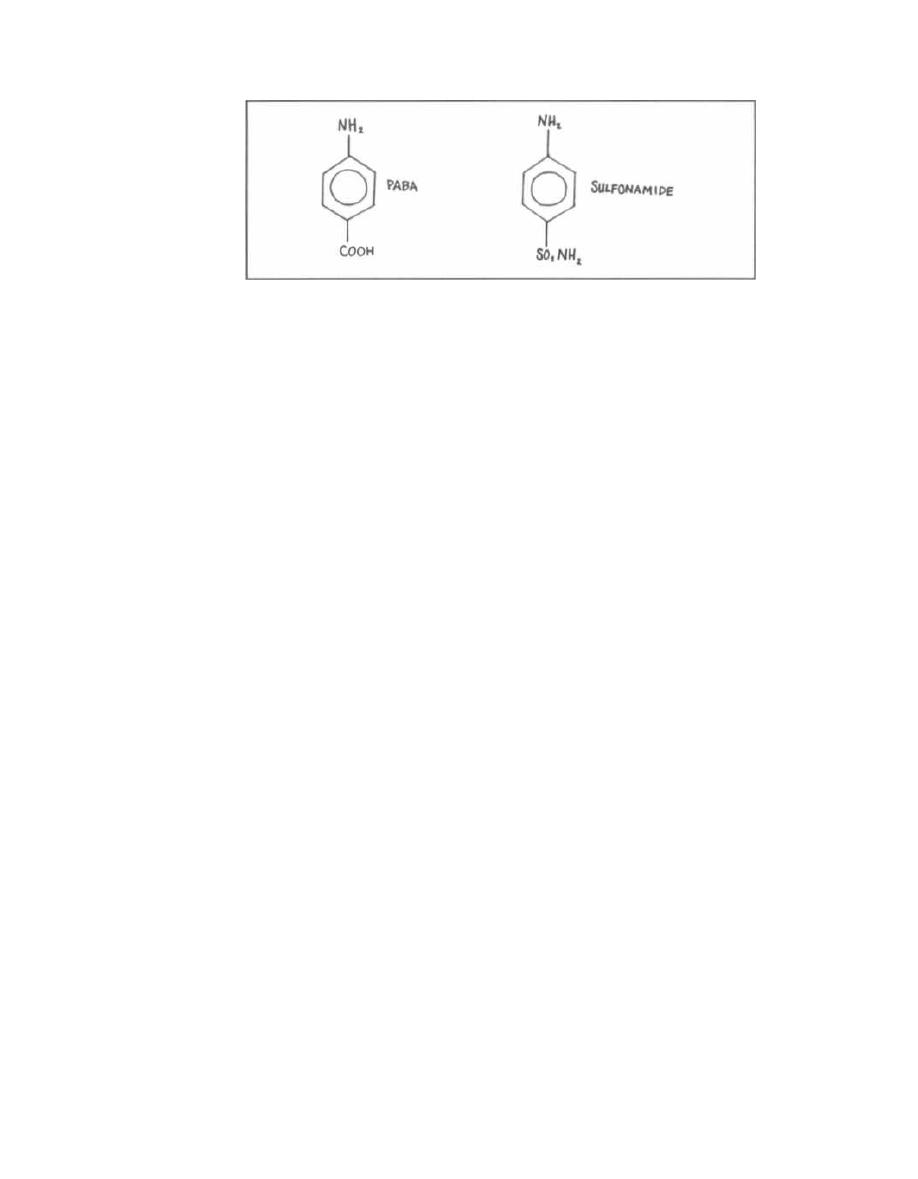
Figure 19-4
TH4 gives up carbons to form purines and other meta-
bolic building blocks. After giving up a carbon, it be-
comes dihydrofolate (TH2) and must be reduced back
to TH4 by the enzyme dihydrofolate reductase.
Trimethoprim looks like the dihydrofolate reductase of
bacteria and competitively inhibits this reduction. This
inhibits bacterial DNA formation.
The big picture here is that trimethoprim
(TMP) and sulfamethoxazole (SMX) act synergis-
tically to kill many gram-positive and gram-nega-
tive bacteria. They both inhibit TH4 production
but at different steps.
Pharmacokinetics
Oral absorption: Just imagine eating a big chunk of
rotten egg which smells like sulfur.
Excretion: Because it is excreted in the urine, this is
a good drug for urinary tract infections. To remember
this, think of the pungent smell of sulfur, that rotten egg
smell, and then think of the pungent smell of urine
when you walk by a Porta-Potti at a construction site.
Make this smell association, and you won't forget.
Adverse Effects
Adverse effects are rare in persons without AIDS.
They include nausea, vomiting, diarrhea, and skin
rashes (drug eruptions). Approximately half of persons
with AIDS develop adverse effects on TMP/SMX, in-
cluding skin rashes and bone marrow suppression.
Giving TMP/SULFA to a patient on warfarin blood thin-
ner is very dangerous! This drug interaction increases war-
farin levels, resulting in a high risk of bleeding.
Clinical Uses
TMP/SMX has no anaerobic coverage, but does have
a wide gram-negative and gram-positive coverage (and
even covers some Protozoans). Study the following
mnemonic TMP SMX:
CHAPTER 19. MISCELLANEOUS ANTIBIOTICS
142
T (Tree): Respiratory tree. TMP/SMX covers Strepto-
coccus pneumoniae and Haemophilus influenzae. It is
good for otitis media, sinusitis, bronchitis, and pneumo-
nia, which are frequently caused by these bugs.
M (Mouth): Gastrointestinal tract. TMP/SMX covers
gram-negatives that cause diarrhea such as Shigella,
Salmonella, and Escherichia coli.
P (PEE): Genitourinary tract. TMP/SMX covers uri-
nary tract infections, prostatitis, and urethritis caused
by the Enterics ( Escherichia coli and clan).
SMX (Syndrome): AIDS. TMP/SMX covers Pneumo-
cystis carinii pneumonia (PCP). It is given to prevent
PCP when CD4+ T-cell counts drop below 200-250.
More than 60% of PCP infections are being prevented
with this prophylactic intervention! It is also given in-
travenously in high doses for active pneumonia.
In addition to Pneumocystis carinii, other protozoans
covered by TMP/SMX are Toxoplasma gondii and
Isospora belli.
Fig. 19-5.
Summary of the miscellaneous antibiotics.
References
Fraser KL, Grossman RF. What new antibiotics to offer in the
outpatient setting. Seminars on Respiratory Infections.
1998;13:24-35.
Frieden TR, Mangi RJ. Inappropriate use of oral ciprofloxacin.
JAMA 264:1438-1440,1990.
Gilbert DN, Moellering RC, Sande MA. The Sanford Guide to
Antimicrobial Therapy 1998. 28th Edition. Antimicrobial
Therapy Inc., Dallas Texas 1998.
Martin SJ, Meyer JM, et al. Levofloxacin and Sparfloxacin: New
quinolone antibiotics. Ann Pharmacother 1998;32: 320-36.
Recommended Review Articles:
Smilack JD. Trimethoprim-Sulfamethoxazole. Symposium on
Antimicrobial Agents. Mayo Clin Proc 74:730-734, 1999.
Walker RC. The Fluoroquinolones. Symposium on Antimicro-
bial Agents. Mayo Clin Proc 74:1030-1037, 1999.
Wilhelm MP, Estes L. Vancomycin. Symposium on Anti-
microbial Agents, Mayo. Clin Proc 74:928-935, 1999.

Figure 19-5
MISCELLANEOUS ANTIBIOTICS
M. Gladwin and B. Trattler,
Clinical Microbiology
Made Ridiculously Simple
OMedMaster

As "budding" doctors in the modern world of AIDS, or-
gan transplantation, and modern chemotherapy, you
will treat an unprecedented number of immunocompro-
mised patients. With their lowered cell-mediated im-
munity, there is a dramatic increase in the incidence of
virtually every fungal infection! You will commonly see
fungi that used to be exceedingly rare.
Fungi are eucaryotic cells, which lack chlorophyll, so
they cannot generate energy through photosynthesis.
They do require an aerobic environment. After dis-
cussing the following crucial terms, we will discuss the
categories of fungi pathogenic to humans.
Yeast: Unicellular growth form of fungi. These
cells can appear spherical to ellipsoidal. Yeast repro-
duce by budding. When buds do not separate, they can
form long chains of yeast cells, which are called
pseudohyphae. Yeast reproduce at a slower rate than
bacteria.
Hyphae: Threadlike, branching, cylindrical, tubules
composed of fungal cells attached end to end. These
grow by extending in length from the tips of the tubules.
Molds (also called Mycelia): Multicellular colonies
composed of clumps of intertwined branching hyphae.
Molds grow by longitudinal extension and produce
spores.
Spores: The reproducing bodies of molds. Spores are
rarely seen in skin scrapings.
Dimorphic fungi: Fungi that can grow as either a
yeast or mold, depending on environmental conditions
and temperature (usually growing as a yeast at body
temperatures).
Saprophytes: Fungi that live in and utilize organic
matter (soil, rotten vegetation) as an energy source.
FUNGAL MORPHOLOGY
Certain morphologic characteristics serve as viru-
lence factors as well as targets for antifungal antibi-
otics.
Cell membrane: The bilayered cell membrane is the
innermost layer around the fungal cytoplasm. It con-
tains sterols (sterols are also found in the cell mem-
branes of humans as well as the bacteria
Mycoplasma).
Ergosterol is the essential sterol in fungi, while cho-
lesterol is the essential sterol in humans. The antibi-
otics amphotericin B and nystatin bind to ergosterol
and punch holes in the fungal cell membrane, while ke-
toconazole inhibits ergosterol synthesis.
PART 2. FUNGI
CHAPTER 20. THE FUNGI
144
Cell wall: Surrounding the cell membrane is the cell
wall, composed mostly of carbohydrate with some pro-
tein. Fungal cell walls are potent antigens to the human
immune system.
Capsule: This is a polysaccharide coating that sur-
rounds the cell wall. This antiphagocytic virulence fac-
tor is employed
by Cryptococcus
neoformans.
The
capsule can be visualized with the India ink stain.
Fig. 20-1. It is helpful to organize the human fungal
diseases by the depth of the skin that they infect.
SUPERFICIAL FUNGAL INFECTIONS
Pityriasis versicolor and tinea nigra are ex-
tremely superficial fungus infections, whose
pri-
mary manifestation is pigment change of the skin.
Neither of these cause symptoms and will only come
to your attention because skin pigment change is
noted!!! Both are named for their respective skin
manifestations:
Pityriasis versicolor ( multicolored)
Tinea nigra (black colored)
1) Pityriasis versicolor (also called tinea versi
-
color) is a chronic superficial fungal infection which
leads to hypopigmented or hyperpigmented patches
on the skin. With sunlight exposure the skin around the
patches will tan, but the patches will remain white. This
infection is caused
by
Malassezia furfur.
2) Tinea nigra is a superficial fungal infection that
causes dark brown to black painless patches on the soles
of the hands and feet. This infection is caused
by Ex-
ophiala werneckii.
Diagnosis of both infections is based on microscopic
examination of skin scrapings, mixed on a slide with
potassium hydroxide (KOH). This will reveal hyphae
and spherical yeast, as the KOH digests nonfungal
debris.
Malassezia
can look like spaghetti (hyphae) with
meatballs (spherical yeast).
Treatment of both consists of spreading dandruff
shampoo containing selenium sulfide over the skin.
This is an inexpensive and effective treatment. The top-
ical antifungal imidazoles can also be used.
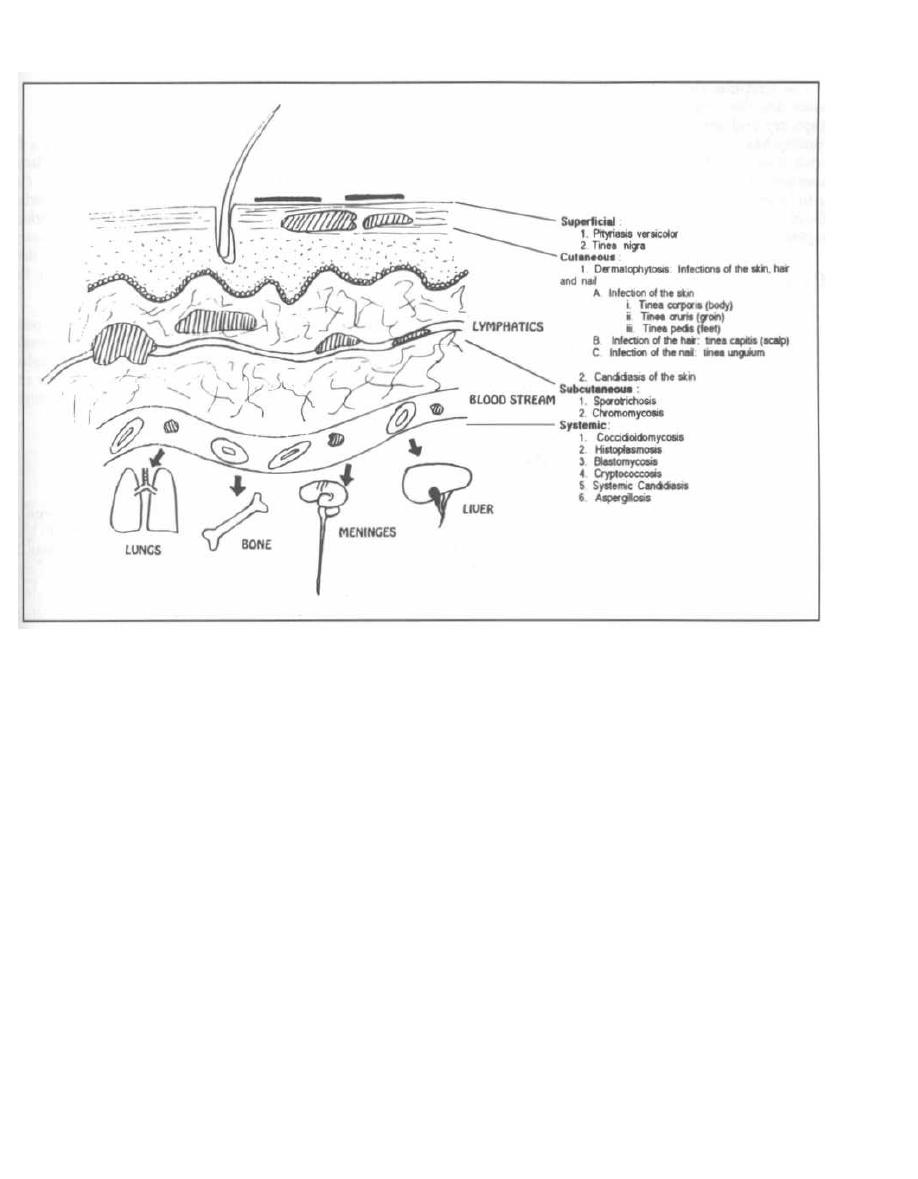
CHAPTER 20.
THE FUNGI
Figure 20-1
CUTANEOUS FUNGAL INFECTIONS of
the SKIN, HAIR, and NAILS
The Dermatophytoses
Dermatophytoses are a category of cutaneous fungal
infections caused by more than 30 species of fungi. The
dermatophytic fungi live in the dead, horny layer of the
skin, hair, and nails (cutaneous layer seen in Fig. 20-1).
These fungi secrete an enzyme called keratinase, which
digests keratin. Since keratin is the primary structural
protein of skin, nails, and hair, the digestion of keratin
manifests as scaling of the skin, loss of hair, and crum-
bling of the nails.
The common dermatophytes include
Microsporum,
Trichophyton,
and
Epidermophyton.
1) Tinea corporis (body): Following invasion of the
horny layer of the skin, the fungi spread, forming a ring
shape with a red, raised border. This expanding raised
red border represents areas of active inflammation with
a healing center. This is appropriately called ringworm,
since it looks like a ring-shaped worm under the skin.
145
2) Tinea cruris (jock itch): Patients develop itchy
red patches on the groin and scrotum.
3) Tinea pedis (athlete's foot): This infection com-
monly begins between the toes, and causes cracking and
peeling of the skin. Infection requires warmth and mois-
ture, so it only occurs in those wearing shoes.
4) Tinea capitis (scalp): This condition primarily
occurs in children. The infecting organisms grow in the
hair and scalp, resulting in scaly red lesions with loss of
hair. The infection appears as an expanding ring.
5) Tinea unguium (onychomycosis) (nails): The
nails are thickened, discolored, and brittle.
To diagnose a dermatophyte infection:
1) Dissolve skin scrapings in potassium hydroxide
(KOH). The KOH digests the keratin. Microscopic ex-
amination will reveal branched hyphae.
2) Direct examination of hair and skin with Wood's
light (ultraviolet light at a wavelength of 365nm). Cer-
tain species of
Microsporum
will fluoresce a brilliant
green.

The first-line drugs for treatment of dermatophy-
toses are the topical imidazoles. The skin should be
kept dry and exposed to the drying effects of the air
(nudity has its advantages!!). Oral griseofulvin is used
with tinea capitis and tinea unguium. Griseofulvin
becomes incorporated into the newly synthesized ker-
atin layers, inhibiting the growth of fungi. So the skin
fungi is cleared only after the old keratin has been
replaced.
Candida albicans
The last type of cutaneous fungal infection is caused
by Candida albicans. Candida can infect the mouth
(oral thrush), groin (diaper rash), and the vagina
(Can-
dida
vaginitis). It can also cause opportunistic systemic
infections. All these infections will be discussed in more
detail later in this chapter (page 150).
SUBCUTANEOUS FUNGAL
INFECTIONS
Subcutaneous fungal infections gain entrance to the
body following trauma to the skin. They usually remain
localized to the subcutaneous tissue or spread along
lymphatics to local nodes. These fungi are normal soil
inhabitants and are of low virulence.
Figure 20-2
CHAPTER 20. THE FUNGI
146
Sporothrix schenckii
(Sporotrichosis)
Fig. 20-2. Spore tricks. Sporothrix schenckii is a di-
morphic fungi commonly found in soil and on plants
(rose thorns and splinters). Sporotrichosis, the dis-
ease, is an occupational hazard for gardeners. Following
a prick by a thorn contaminated with
Sporothrix
schenckii,
a subcutaneous nodule gradually appears.
This nodule becomes necrotic and ulcerates. The ulcer
heals, but new nodules pop up nearby and along the
lymphatic tracts up the arm.
Microscopic examination of this fungus reveals yeast
cells that reproduce by budding. Culture at 37°C reveals
yeast, while culture at 25°C reveals branching hyphae
(dimorphism). Treat with oral potassium iodide or am-
photericin B. So if you are going to POT roses you might
buy some potassium iodide!
Phialophora
and
Cladosporium
(Chromoblastomycosis)
Fig. 20-3. Visualize a chrome-plated (chromo)
fungi blasting cauliflower warts on the skin to help
you remember the disease Chromoblastomycosis. It
is a subcutaneous infection caused by a variety of cop-
per-colored soil saprophytes
(Phialophora
and Cla-

Figure 20-3
dosporium)
found on rotting wood. Infection occurs fol-
lowing a puncture wound. Initially, a small, violet wart-
like lesion develops. Over months to years,
additional violet-colored wartlike lesions arise nearby.
Clusters of these lesions resemble cauliflower. Skin
scrapings with KOH reveal copper-colored sclerotic
bodies. Treat with itraconazole and local excision.
SYSTEMIC FUNGAL INFECTIONS
Three fungi that cause systemic disease in humans
are
Histoplasma capsulatum, Blastomyces dermati-
tides,
and
Coccidioides immitis.
All 3 are dimorphic
fungi. They grow as mycelial forms, with spores, at
25°C on Sabouraud's agar. At 37°C on blood agar, they
grow in a yeast form. This dimorphism plays a part in
human infection. In their natural habitat (the soil) they
grow as mycelia and release spores into the air. These
spores are inhaled by humans and at the "human tem-
perature" of 37°C they grow as yeast cells.
Geography
Fig. 20-4.
Histoplasma
and Blastomyces are en-
demic to the vast areas that drain into the Mississippi
River. Visualize a fungi pilot firing a rocket that HITS
and BLASTS a hole in the Mississippi River.
Fig. 20-5.
Coccidioides
is endemic to the southwestern
U.S. (Arizona, New Mexico, southern California) and
northern Mexico. Visualize Mr. Fungus as he COCKS
his pistol in the old SOUTHWEST.
CHAPTER 20.
THE FUNGI
14 7
Knowledge of these geographic areas is important
clinically. For example,
Coccidioides
has become the
second most common opportunistic infection in AIDS
patients who have resided in Arizona. So a sick AIDS
patient with a history of previous residence in the
Southwest would raise suspicions.
Mechanism
of
Disease
All 3 fungi have a similar disease mechanism. Notice
the parallels to tuberculosis.
Like
Mycobacterium tuberculosis
the 3 fungi are
acquired by inhalation. However, unlike
Mycobac-
terium tuberculosis,
the fungal infections are inhaled
as a spore form and are never transmitted from per-
son to person. Rather, the spores are aerosolized from
soil, bird droppings, or vegetation. Like
Mycobacterium
tuberculosis,
once inhaled, local infection in the lung
is followed by bloodstream dissemination. In most
infected persons the fungi are destroyed at this point
by the cell-mediated immune system. Antigenic prepa-
rations called coccidioidin and histoplasmin are
like the PPD of
Mycobacterium tuberculosis:
when
injected intradermally in a previously exposed per-
son they yield a delayed type hypersensitivity reac-
tion which results in localized swelling within 24-48
hours.
The 3 fungi have 3 clinical presentations:
1) Asymptomatic: The majority of cases are asymp-
tomatic or mild respiratory illnesses that go unreported.
2) Pneumonia: A mild pneumonia can develop with
fever, cough, and chest X-ray infiltrates. Like tubercu-
Figure 20-5
losis, granulomas with calcifications can follow resolu-
tion of the pneumonia.
A small percentage of persons will develop a severe
pneumonia, and an even smaller group will progress to
a chronic cavitary pneumonia, marked by weight loss,
night sweats, and low-grade fevers, much like a chronic
tuberculosis pneumonia.
3) Disseminated: Rarely, the hematogenously
spread fungi can actually cause disseminated disease,
such as meningitis, bone lytic granulomas, skin granu-
lomas that break down into ulcers, and other organ le-
sions. This disseminated form commonly occurs in the
immunocompromised host.
All 3 are best diagnosed by obtaining a biopsy of the
affected tissue: bronchoscopic biopsy of lung lesions,
CHAPTER 20.
THE FUNGI
Figure 20-4
1
148
skin biopsy, etc. The tissue can be examined with silver
stain for yeast or can be grown on Sabouraud's agar or
blood agar. The tissue is the issue!!!! Skin tests are
not very helpful for diagnosis, as many people have been
previously exposed asymptomatically and will have a
positive test anyway. Serologic tests can be helpful
(complement fixation, latex agglutination).
Acute pulmonary histoplasmosis and coccidioidomy
cosis usually require no treatment, as the infection is
mild. For chronic or disseminated disease, itraconazole
or amphotericin B is often required for months! All
Blas.
tomyces
infections require aggressive amphotericin B or
itraconazole treatment.
Summary of
Histoplasma, Blastomyces,
and
Coccidioides
LIKE TUBERCULOSIS
Inhaled, primary in fection in the lung.
Asymptomatic, mild, severe,
or chronic lung infections.
Lung granulomas, calcifications,
and/or cavitations.
Can disseminate hematogenously
to distant sites.
Skin test like PPD.
UNLIKE TUBERCULOSIS
No person-to-person transmission.
Fungi with spores, NOT acid-fast
bacteria.
Fig. 20-6. To remember that Coccidioides,
Blasto.
myces, and Histoplasma all are inhaled as spores and
cause disease in the lungs, skin, bones, and meninges,
study Cowboy Fungus. He has spore bullets, cocks
his gun, then blasts and hits the lung, skin, bone, and
meninges.
Figure 20-6
Histoplasma capsulatum:
Nonencapsulated despite its name.
Present in bird and bat droppings, so outbreaks of
pneumonia occur when cleaning chicken coops or
spelunking (cave exploring).
Blastomyces dermatitides:
Fungi are isolated from soil and rotten wood.
The rarest systemic fungal infection.
When it does cause infection, it is rarely asympto-
matic or mild. Most cases present as chronic dissemi-
nated disease with weight loss, night sweats, lung
involvement, and skin ulcers. Blastomyces is the hard-
est to get and the hardest to have!
The bLAST to get,
No BLAST to have!!!
Coccidioides immitis:
Commonly causes a mild pneumonia in normal per-
sons in the southwestern U.S.
Common opportunistic infection in AIDS patients
from that area.
Cryptococcus neoformans
(Cryptococcosis)
Cryptococcus neoformans
is a polysaccharide encap-
sulated yeast (not dimorphic) similar to the previous 3
fungi in that it is inhaled into the lungs and the in-
fection is usually asymptomatic. It differs in that the
major manifestation is not pneumonia but rather
meningoencephalitis.
CHAPTER 20.
THE FUNGI
14 9
This fungus is found in nature, especially in pigeon
droppings. Following inhalation and local lung in-
fection, often asymptomatic, the yeast spreads via
the blood to the brain. There is no geographic clus-
tering of infections as with the other systemic my-
coses. Most cases (3/4) occur in immunocompromised
persons. In fact, almost 10% of AIDS patients develop
cryptococcosis.
A subacute to chronic meningitis develops in crypto-
coccosis with headache, nausea, confusion, staggering
gait, and/or cranial nerve deficits. Fever and meningis-
mus can be mild. Cryptococcal meningitis is fatal with-
out treatment, because cerebral edema progresses to
eventual brainstem compression.
Cryptococcus
can also cause pneumonia, skin ulcers,
and bone lesions like the other systemic fungi.
The key to diagnosis is doing a lumbar puncture and
analyzing the cerebrospinal fluid. An India ink stain
shows yeast cells with a surrounding halo, the polysac-
charide capsule. This test is positive half of the time. A
more sensitive test is the cryptococcal antigen test,
which detects cryptococcal polysaccharide antigens.
Culture will confirm the diagnosis.
Fig. 20-7.
AIDS and cryptococcosis.
The usual treatment is with amphotericin B and flucy-
tosine. Persons require treatment for as long as 6
months with serial lumbar punctures to confirm resolu-
tion. AIDS patients may require treatment for life.
CRYPTOCOCCUS
NEOFORMANS
YEAST FROM
C.S.F. STAINED
WITH INDIA INK
Figure 20-7
Candida albicans
( Candidiasis)
As a physician you will see this yeast everywhere. It is
given out like CANDY to humanity: women with vagini-
tis, babies with diaper rash, AIDS patients, and the list
goes on. In the normal host, Candida albicans causes 3
infections that are cutaneous. In the immunocompro-
mised patient, it can cause any of the 3 cutaneous in-
fections, as well as cause invasive systemic disease.
In Normal Hosts
1) Oral thrush: Patches of creamy white exudate
with a reddish base cover the mucous membranes of the
mouth. These are difficult to scrape oft' with a tongue
blade. Swish and spit preparations of nystatin or am-
photericin B, or merely sucking on imidazole candies
will resolve this infection.
2) Vaginitis: There are 20 million cases of "yeast in-
fection" every year in the U. S. alone! Women develop
Candida vaginitis more frequently when taking antibi-
otics, oral contraceptives, or during menses and preg-
nancy. The symptoms are vaginal itching and discharge
(thick copious secretions
wetting the underwear).
Speculum examination reveals inflamed vaginal mu-
CHAPTER 20. THE FUNGI
150
cosa and patches of cottage cheese-appearing white
clumps affixed to the vaginal wall. Imidazole vaginal
suppositories are helpful.
3) Diaper rash: Warm moist areas under diapers
and in adults between skin folds (under breasts for ex
•
ample) can become red and macerated secondary to
Candida i nvasion.
In Immunocompromised Patients
4) Esophagitis: Extension of thrush into the esoph
•
agus causes burning substernal pain worse with swal
lowing. Candida does not infect the esophagus in
i mmune-competent persons!
5) Disseminated: Candida can invade the blood
•
stream and virtually every organ. When systemic can
didiasis is suspected, the retina must be examined with
the ophthalmoscope. Multiple white fluffy candidal
patches occasionally may be visualized. Clinical
Point: Since Candida is normal flora, it is often cul
Lured from the urine, sputum, and stool. These can rep-
resent contaminants. However, isolation from the blood
is never normal and must be respected!!!
Diagnosis is made with KOH preparation of skin
scrapings, or with stains and cultures of biopsied tissue
or blood.
Figure 20-8
Systemic infection requires amphotericin B or the
oral antifungal imidazole called fluconazole.
Aspergillus flavus
(Aspergillosis)
ASPIRATION of
ASPERGILLUS
= ASTHMA
Fig. 20-8.
The spores
of Aspergillus
mold are floating
in the air everywhere. Some persons develop an
asthma-type reaction to these spores. They have a type
1 hypersensitivity reaction (IgE-mediated immediate
allergic reaction) with bronchospasm, increase in IgE
antibodies, and blood eosinophilia. They also manifest a
type 4 reaction (delayed type cell-mediated allergic re-
action) with cell-mediated inflammation and lung infil-
trates.
Systemic corticosteroids
are
an effective
treatment.
Fig. 20-9.
Persons with lung cavitations from tuber,
culosis or malignancies can grow an aspergillus fungal
ball in the cavity, called an aspergilloma. This ball can
be large (as big as a golf ball) and require surgical re-
moval.
I mmunocompromised hosts can develop invasive
pneumonias and disseminated disease. Bloody sputum
may occur, due to blood vessel wall invasion by As-
pergillus
hyphae.
Aspergillus flavus
and other fungi produce toxins
that cause liver damage and liver cancer. These toxins
are called mycotoxins. The toxin produced by As-
pergillus flavus
is called the aflatoxin. This has world-
wide significance since
Aspergillus
grows ubiquitously,
contaminating peanuts, grains, and rice. The fact that
half of the cancers south of the Sahara desert in Africa
are liver cancers and 40% of screened foods contain afla-
toxins suggests that this is a real threat.
CHAPTER 20.
THE FUNGI
15 1
Figure 20-9
THE FUNGI-LIKE BACTERIA:
ACTINOMYCETES and NOCARDIA
Fig. 20-10.
Actinomycetes are bacteria acting like
fungi. These are procaryotic organisms and are truly
bacteria. They are discussed here because they fre-
quently grow in the form of mycelia and are water and
soil saprophytes. The 2 that cause human disease are
Figure 20-10

Actinomyces and Nocardia, both of which are gram-
positive rods.
Actinomyces Israelii
There are 4 concepts you should know about this
organism:
1) It is a gram-positive, beaded, filamentous anaero-
bic organism that grows as normal flora in the mouth
and GI tract.
2) It causes eroding abscesses following trauma to
the mucous membranes of the mouth or GI tract. The in-
fection is named according to the area of the body
through which the abscess erodes: cervicofacial
actinomycosis, abdominal actinomycosis, and tho-
racic actinomycosis.
3) When examined under the microscope, the pus
draining from the abscess reveals yellow granules,
called sulfur granules. These are not composed of
sulfur but of microcolonies of Actinomyces and cellular
debris.
4) Treatment of this gram-positive bacterium is with
penicillin G and surgical drainage.
Note: Actinomyces and Nocardia are both filamen-
tors, beaded, branching gram-positive organisms, but
CHAPTER 20. THE FUNGI
152
only Actinomyces forms sulfer granules and only
No
cardia is acid-fast.
Nocardia asteroides
Nocardia forms weakly gram-positive, partially
acid-fast beaded branching thin filaments! It is not
considered normal flora. Infections with Nocardia are
frequently misdiagnosed as tuberculosis because it is
acid-fast and it causes the same disease process. Like
Mycobacterium tuberculosis, Nocardia is inhaled and
grows in the lung to produce lung abscesses and cavita-
tions. Erosion into the pleural space can occur, as well
as blood-bourne dissemination, resulting in abscesses in
the brain and other organs. Immunocompromised pa-
tients, especially those taking steroids, are particularly
at risk for Nocardia infection. Treatment is with
trimethoprim and sulfamethoxazole.
Treatment ofActinomyces and Nocardia is a SNAP!
Sulfa for
Nocardia
Actinomyces give
Penicillin
Fig. 20-11. Summary of the fungi.
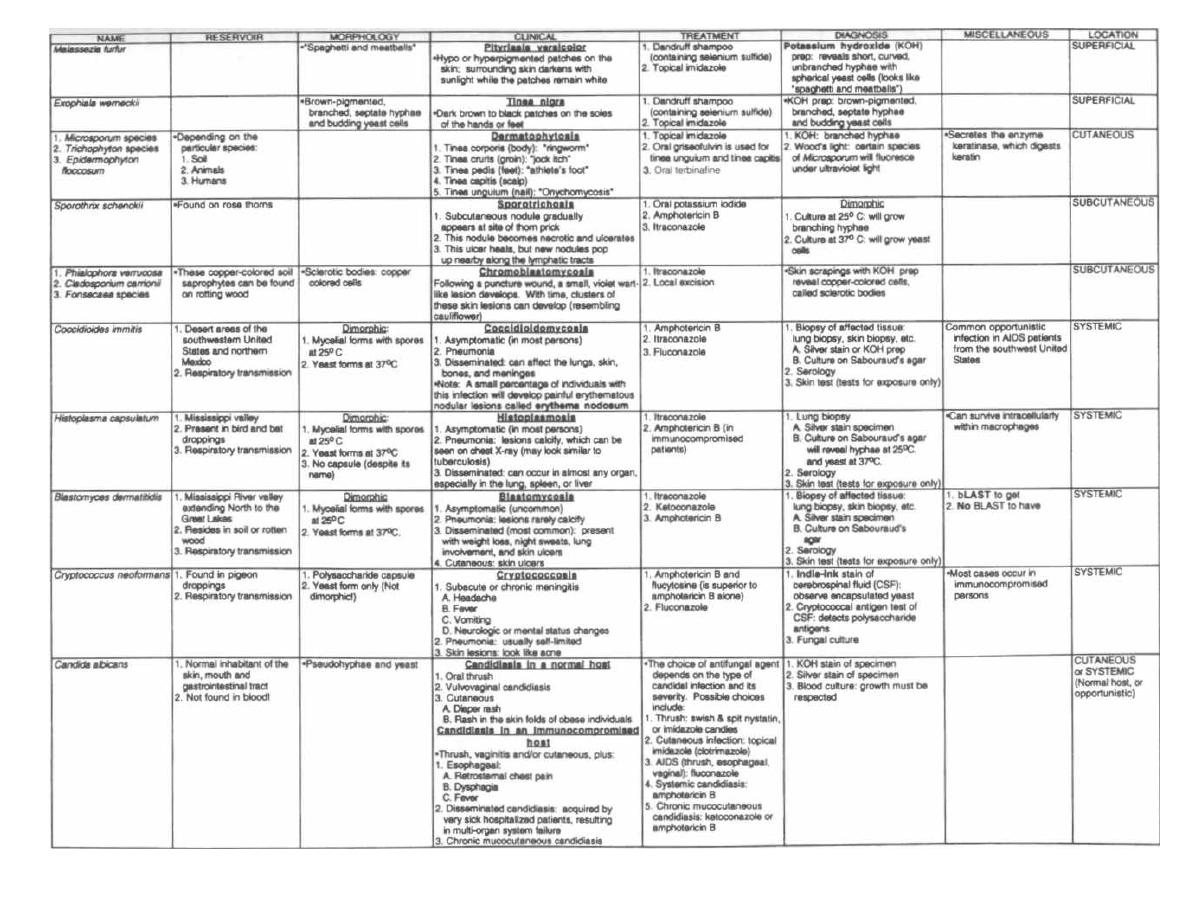
Figure 20-11
FUNGI
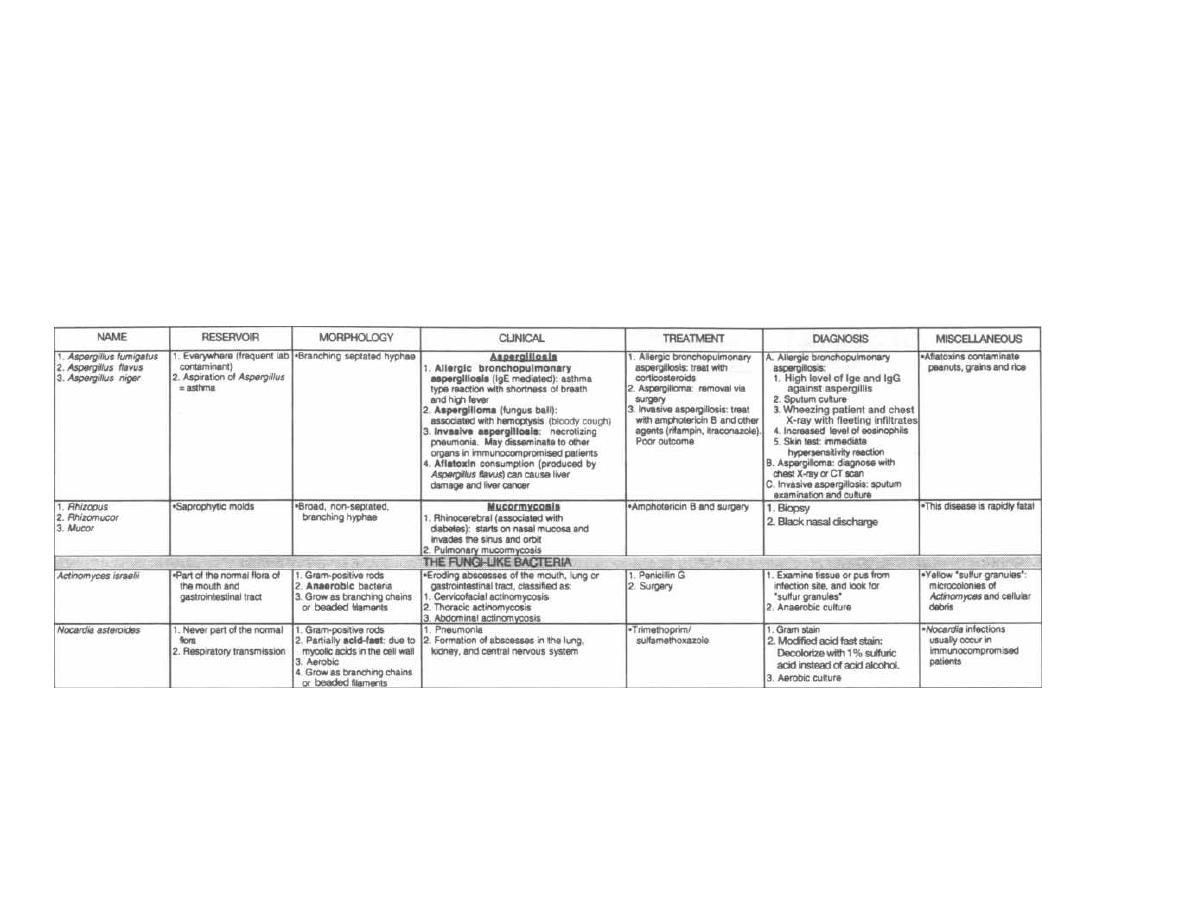
Figure 20-11 (continued)
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
C~MedMaster

CHAPTER 21. ANTIFUNGAL ANTIBIOTICS
The number of fungal infections has risen dramatically
with the increase in patients who are immunocompro-
mised from AIDS, chemotherapeutic drugs, and organ
transplant immunosuppressive drugs. For this reason
you will frequently use the handful of antifungal an-
tibiotics available.
Ergosterol is a vital part of the cell membranes of
fungi but is not found in the cell membranes of humans,
which contain cholesterol. Most antifungal agents bind
more avidly to ergosterol than to cholesterol, thus more
selectively damaging fungal cells than human cells. By
binding to or inhibiting ergosterol synthesis, they in-
crease the permeability of the cell membranes, causing
cell lysis.
We can divide antifungal agents into 3 groups:
1) Antifungal agents that are used for serious sys-
temic infections:
Amphotericin B, the grandfather of antifungal
agents. This drug covers almost all medically impor-
tant fungi but must be given intravenously (not ab-
sorbed orally) and causes many side effects. It may
also be given intrathecally (into the cerebrospinal
fluid).
Itraconazole, given orally, has now proven use-
ful for many of these infections.
2) Antifungal agents that are used for less serious
systemic infections:
The oral azole drugs. The prototype is keto-
conazole. There are now new agents in this class,
called fluconazole and itraconazole ( mentioned
above).
3) Antifungal agents that are used for superficial
fungal infections:
Griseofulvin (taken orally) and the many
topical antifungal agents such as nystatin
and the azole drugs (clotrimazole and
miconazole).
Amphotericin B
The classic antifungal antibiotic is amphotericin B.
Most species of fungi are susceptible to it, and although
it has many side effects, it remains the drug of choice for
most serious systemic fungal infections:
Systemic Candida i nfections.
Cryptococcal meningitis: used in combination with
flucytosine.
Severe pneumonia and extrapulmonary Blastomyco-
sis, Histoplasmosis, and Coccidioidomycosis.
Invasive Aspergillosis.
Invasive Sporotrichosis.
Mucormycosis
15,5
Adverse Effects
On the wards this drug is referred to as am-
photerrible and Awfultericin because of its nu-
merous adverse effects:
1) Renal toxicity: (Poor Mr. Kidney!) There is a
dose-dependent azotemia (increase in BUN and creati-
nine reflecting kidney damage) in most patients taking
this drug. This is reversible if the drug is stopped. The
creatinine level must be followed closely, and if it be-
comes too high (creatinine > 3), the dosage may have to
be lowered, terminated, or switched to alternate day
regimens.
2) Acute febrile reaction: A shaking chill (rigors)
with fever occurs in some people after IV infusion. This
is a common side effect.
3) Anemia.
4) Inflammation of the vein ( phlebitis) at the IV site.
These side effects are important because they are
very common. In fact, when amphotericin B is given in
the hospital, it is usually given with aspirin or aceta-
minophen to prevent the febrile reaction. Daily BUN
and creatinine levels are drawn to monitor kidney func-
tion. You can see that these side effects are important
in day-to-day clinical management!
Fig. 21-1. Properties of amphotericin B, the "am-
phibian terrorist": i ntravenous drug delivery; fung-
icidal by binding to ergosterol in the fungal cell mem-
brane, causing membrane disruption and osmotic lysis
of the cell; nephrotoxicity.
To speed amphotericin's travel through the kidneys
and decrease renal toxicity, hydration with normal
saline is used commonly with traditional amphotericin
B. This hydration is generally not required with the
newer preparations of amphotericin B. Electrolyte re-
placement is another important adjunct of ampho-
tericin therapy because amphotericin causes increases
in urinary excretion of potassium, magnesium and
bicarbonate.
New preparations of amphotericin B are now available
that add different lipids (fats!) to the traditional (old fash-
ion) amphotericin B deoxycholate. The addition of the
lipid decreases the nephrotoxicity of the drug, making it
less Amphoterrible.
Amphotericin B colloidal dispersion (ABCD:
Amphocil): Ampho B + cholesterol sulfate. Rigors still
occur but nephrotoxicity is reduced.
Amphotericin B lipid complex (ABLC): Ampho
B + dimyristoylphosphatidylglycerols and dimyris-
toylphosphatidylcholines. Rigors still occur but there is
less nephrotoxicity.

Figure 21-1
Liposomal amphotericin B (Ambisome): A unil-
amellar liposome containing a mixture of 1 molecule of
amphotericin B surrounded by a coating of nine mole-
cules of lipid (soy lecithin, cholesterol, and dis-
tearoylphosphatidylglycerol), like a coated jaw breaker.
There is little nephrotoxicity or rigors.
Some hospitals make their own concoction by adding
amphotericin B deoxycholate to Intralipid (parenteral
fat for intravenous feeding) in a mixture of 1-2 mg am-
photericin B per ml lipid. Less nephotoxicity is seen, but
once again we do not yet know enough about antifun-
gal efficacy.
Flucytosine
Flucytosine is rarely used alone because of rapid de-
velopment of resistance. Think of it as the tag team
CHAPTER 21.
ANTIFUNGAL ANTIBIOTICS
156
wrestling partner of amphotericin B. Amphotericin B
busts holes in the cell membranes and flucytosine en-
ters and inhibits DNA/RNA synthesis.
Most fungi are resistant to flucytosine, but
Crypto-
coccus
and
Candida
are the exceptions. Flucytosine use
is mostly limited to the treatment of cryptococcal
meningitis, in conjunction with Amphotericin B.
Adverse Effects
1) Bone marrow depression, resulting in leukope-
nia and thrombocytopenia. Remember that most an-
timetabolite type drugs will do this (methotrexate, sulfa
drugs, 5-fluorouracil, etc.).
2) Nausea, vomiting, diarrhea. This again is com-
mon with the antimetabolites, such as the chemothera-
peutic drugs.

I
The reason for these adverse effects is that the drugs
damage DNA during its formation in rapidly dividing
cells such as bone marrow and GI epithelial cells.
The Azole Family
The azole family may be classified into 2 groups of
drugs: the imidazoles and the triazoles.
IMIDAZOLES
TRIAZOLES
Ketoconazole
Fluconazole
Miconazole
Itraconazole
Clotrimazole
Voriconazole
The azoles inhibit the cytochrome P-450 enzyme sys-
tem, which is involved in ergosterol synthesis. The de-
pletion of ergosterol disrupts the permeability of the
fungal cell membrane.
These drugs are active against a broad spectrum of
fungi.
Clotrimazole and miconazole are too toxic for sys-
temic use and for this reason, are primarily used for
topical fungal infections, including pityriasis versi-
color, cutaneous candidiasis, and the dermatophytosis
(tinea pedis, corporis, etc.). Clotrimazole troches (like
candies) are sucked to treat oral
Candida
(thrush), and
clotri mazole
vaginal suppositories treat
Candida
vaginitis.
Ketoconazole, fluconazole, and itraconazole are
tolerated orally and have many important uses for sys-
temic fungal infections.
Ketoconazole
Ketoconazole, one of the imidazoles, is the drug of
choice for chronic mucocutaneous candidiasis
( Can-
dida
on every surface). It is NOT used for systemic
candidiasis (amphotericin B, remember?). Ketocona-
zole is currently not used for the treatment of systemic
fungal infections. The safer, more efficacious, oral itra-
conazole and old faithful, amphotericin B, are the first
line drugs.
Adverse Effects
1) GI: Nausea, vomiting, and anorexia, all common.
2) Hepatotoxicity: This is usually seen as a tempo-
rary rise of hepatic enzymes but on rare occasions can lead
tohepatic necrosis. Follow enzymes when on this drug.
3) Inhibition of testosterone synthesis: Keto-
conazole inhibits the cytochrome P-450 system, which is
important in testosterone synthesis. The result is gy-
necomastia, impotence, decreased sex drive (libido), and
decreased sperm production.
4) Adrenal suppression.
CHAPTER 21.
ANTIFUNGAL ANTIBIOTICS
157
Fluconazole
Fluconazole is one of the triazoles; it is less toxic and
has broader antifungal coverage than ketoconazole.
Like ketoconazole it is used for cutaneous
Candida
in-
fections but it is also used as a second-line agent behind
amphotericin B for systemic candidiasis and cryptococ-
cal meningitis. In AIDS patients who have had crypto-
coccal meningitis, maintenance with fluconazole will
prevent relapses.
The big picture with fluconazole is that it kills
Can-
dida albicans
very well:
1) Studies comparing it to amphotericin B in the
treatment of systemic
Candida albicans
infection (in
non-neutropenic patients) demonstrated equivalent
effcancy.
2) A single dose of fluconazole very effectively clears
candida vaginitis.
Itraconazole
This triazole is becoming the next amphotericin B but
in an oral formulation without the many amphoterrible
side effects!!!
Itraconazole is now used as first-line treatment
for chromoblastomycosis, histoplasmosis, coccidioido-
mycosis, blastomycosis, and possibly for invasive
aspergillosis. The main problem with this drug is
poor oral absorption. Taking it with acid drinks
such as orange juice or colas enhances absorption
( need low pH). A new IV formulation has also been de-
veloped to avoid poor absorption.
Voriconazole
Voriconazole has a Voracious appetite for fungi!!!
Voriconazole is a promising triazole antifungal. Al-
though more clinical experience is needed, data are suf-
ficient to support its future use in patients with invasive
aspergillosis who have failed to respond to agents of
choice (i.e. conventional or liposomal amphotericin B,
itraconazole).
Other Antifungal Drugs
Nystatin
Nystatin, like amphotericin B, binds to ergosterol, in-
creasing the permeability of the cell membrane and
causing cell lysis.
Think "Nasty Nystatin" because this drug is too
toxic to take parenterally (intravenously). It is only
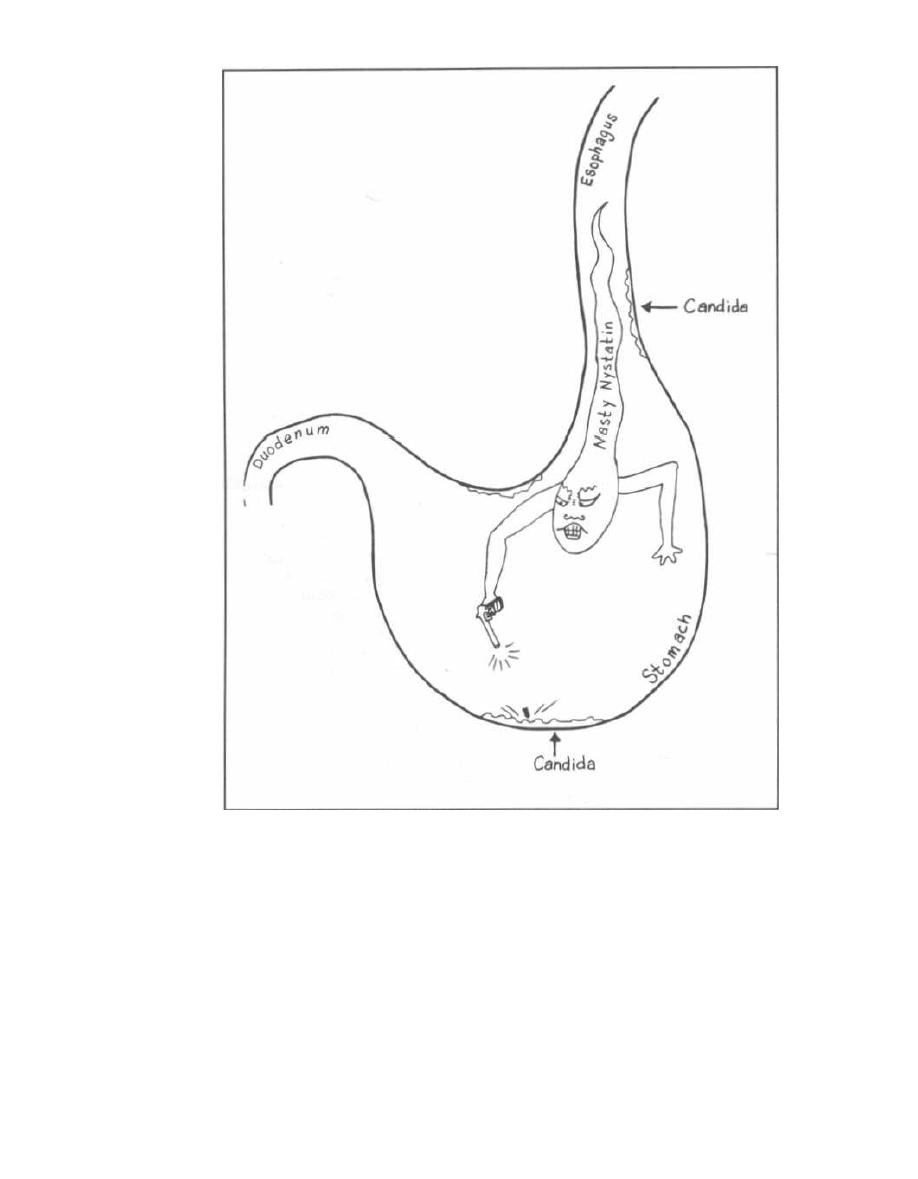
Figure 21-2
used topically on the skin and mucous membranes.
Also, since it is not absorbed from the gastrointestinal
tract, oral nystatin can be used to treat oral and
esophageal infections with yeast or fungi. You will or-
der nystatin on the wards as Nystatin, Swish and
Swallow for treatment of oral, esophageal, and gas-
tric candidiasis. It is also given topically for vaginal
candidiasis.
Fig. 21-2. Nasty Nystatin cruises down the esopha-
gus killing fungi on the wall of the esophagus. In one
end and out the other!
CHAPTER 21.
ANTIFUNGAL ANTIBIOTICS
158
Griseofulvin
Fig. 21-3. Visualize griseofulvin as a greasy ful
•
cram used to lever the dermatophyte plaques off the
skin. It inhibits fungal growth by disrupting spindle for
•
mation, thus preventing mitosis. Note the worker peel
•
ing fungus off your "toe,"sis.
Griseofulvin deposits in keratin precursor cells in
the skin, hair, and nails, where it inhibits the growth
of fungi in those cells. Note that it does not kill the
fungi; it just inhibits their growth (static rather than
cidal). The uninfected drug-infiltrated keratin precur
•
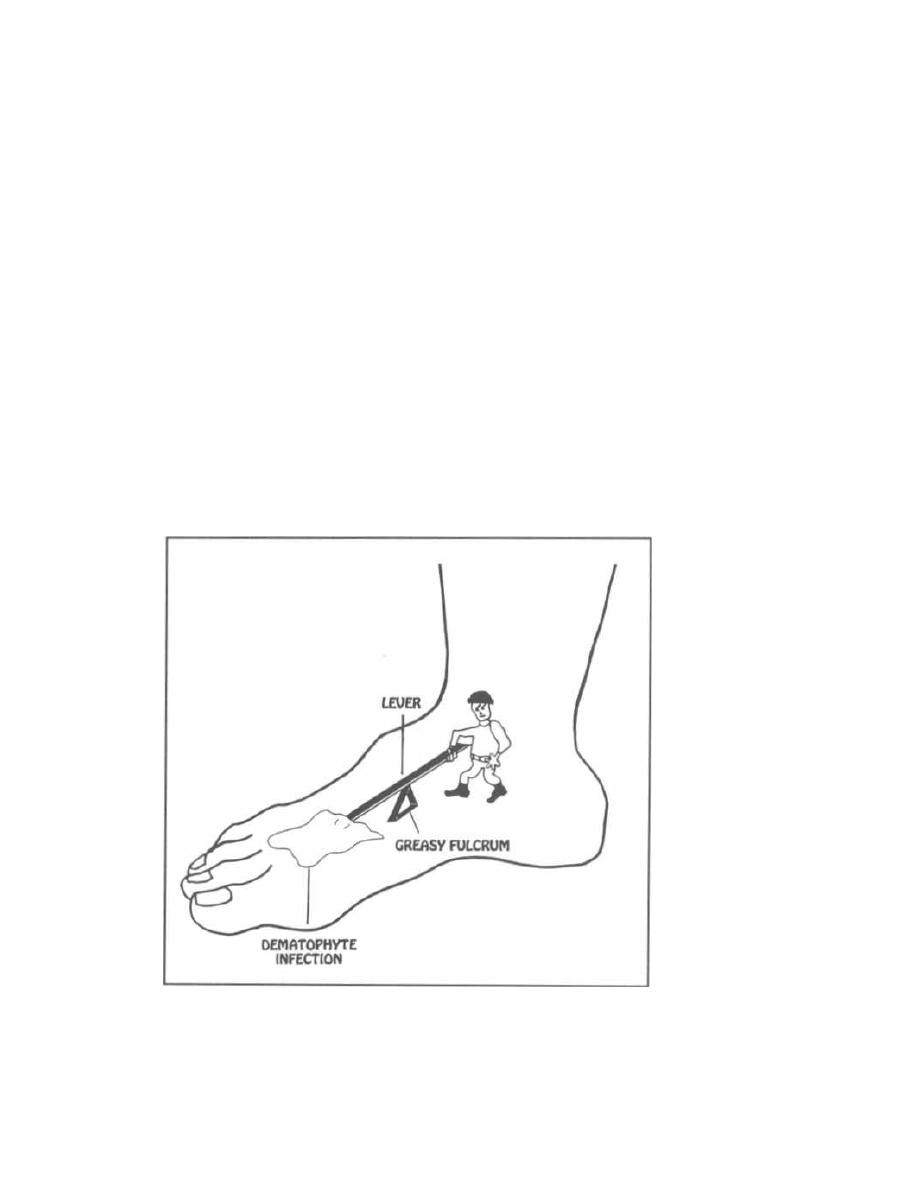
sor cells mature and move outward toward the kera-
tinized layer. As the older, infected cells fall off with
normal cell turnover, this translates into a slow cure
of skin fungus.
Adverse effects of griseofulvin are uncommon. They
include headache, nausea, vomiting, photosensitivity,
and mental confusion, in addition to bone marrow sup-
pression (leukopenia and neutropenia).
Potassium Iodide
Potassium iodide is used to treat sporotrichosis. Re-
member that you get sporotrichosis from pricking your
finger in the garden. "You get Sporotrichosis while Pot-
ting plants." If the infection becomes systemic, ampho-
tericin B or itraconazole is better.
Fig. 21-4.
Summary of the anti-fungal drugs.
Terbinafine
Terbinafine is a new oral fungicidal agent that
blocks fungal cell wall synthesis. It blocks ergosterol
Figure 21-3
CHAPTER
21. ANTIFUNGAL ANTIBIOTICS
159
synthesis by inhibiting the formation of squalene epox-
ide from squalene. Terbinafine tends to accumulate
i n nails, and is therefore useful for tinea unguium
(onychomycosis). It also appears useful in the treat-
ment of tinea pedis, tinea capitis, and tinea corporis.
Since it is not metabolized by the cytochrome p450 sys-
tem (as do the azole antifungals), there is little poten-
tial for drug-drug interactions.
Reference
Bennett JE. Antifungal Agents. In: Mandell GL. Bennett JE,
Dolin R, eds. Principles and Practice of Infectious Diseases.
4th edition. New York: Churchill Livingstone 1995;
401-410.
Jackson CA, et al. Drug therapy: Oral azole drugs as systemic
antifungal therapy. N Engl J Med 1994;330:263-272.
Sanford JP, Gilbert DN, et al. Guide to antimicrobial therapy
1994. Antimicrobial Therapy,
m c,
Dallas Texas; 1994.
Recommended Review Articles:
Terrell CL. Antifungal agents. Mayo Clin Proc 1999:74:78-100.
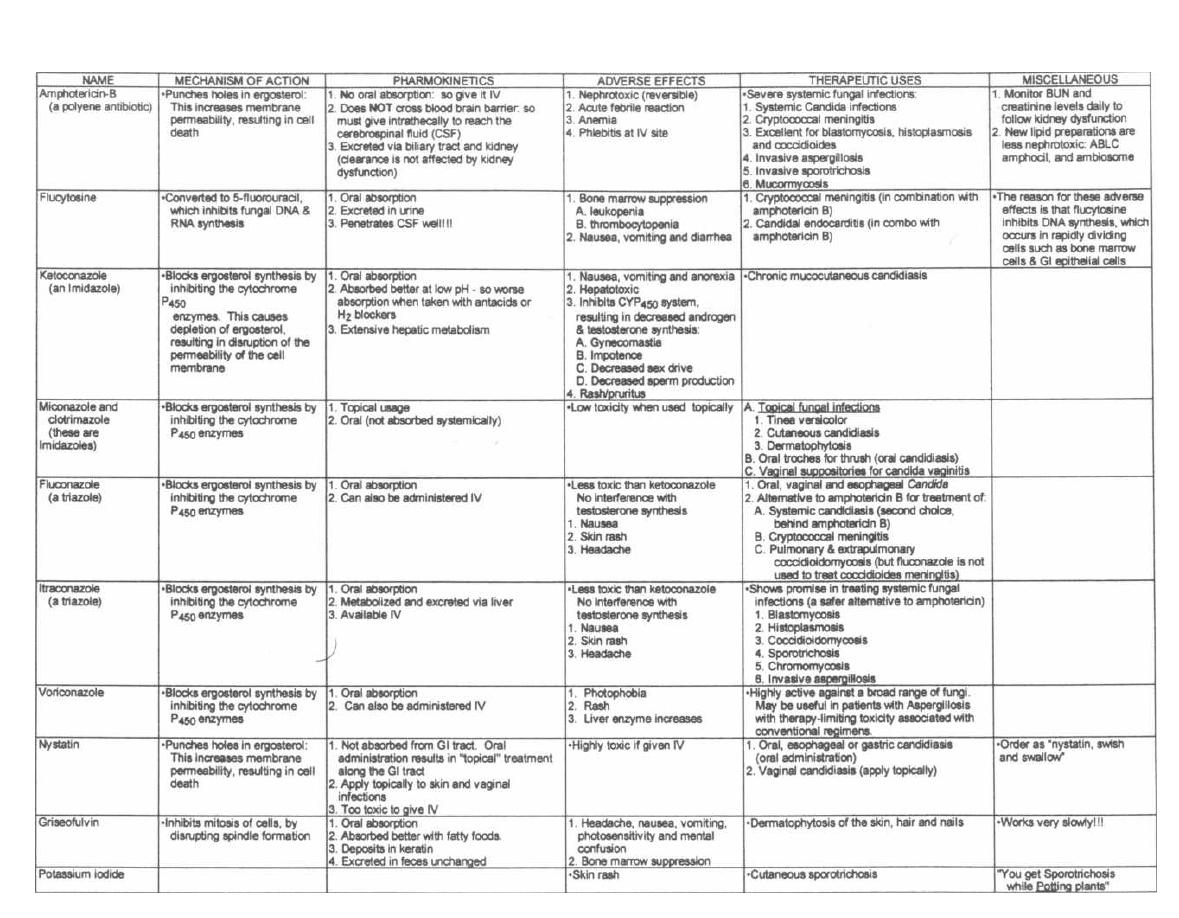
Figure 21-4 ANTI-FUNGAL DRUGS
M. Gladwin and B. Trattler. Clinical Microbiology Made Ridiculously Simple WedMester

Figure 22-1
Fig. 22-1. Imagine that a virus is a specially designed
military spaceship with a super-resistant outer shell.
However, the engineers forgot to build a fuel tank for
their spaceship. It must therefore drift around until it
encounters a satisfactory planet. When it lands, it
transfers the military cargo off the ship. This cargo is
designed to take control of all factories and then instruct
the factories to begin building copies of the original
spacecraft (without fuel tanks). When the replica ships
have been constructed, they will depart in mass, leaving
the planet in complete ruins.
Chapter 22.
PART 3. VIRUSES
VIRAL REPLICATION and TAXONOMY
161
Viruses have these unique characteristics:
1) They are energy-less. They float around until they
come in contact with an appropriate cell.
2) They are basic life forms composed of a protein
coat, called a capsid, that surrounds genetic material.
Viruses do not have organelles or ribosomes. Certain
viruses are further enclosed by an external lipid bilayer
membrane that surrounds the capsid and may contain
glycoproteins. Some viruses also carry some structural
proteins and enzymes inside their capsid.
3) The genetic material is either DNA or RNA.
Never both!! The genetic material contains instruc-
tions to make millions of clones of the original virus.
4) Replication of the genetic material occurs when
the virus takes control of the host cell's synthetic ma-
chinery. Viruses contain all of the genetic information,
but not the enzymes, needed to build millions of replicas
of the original virus.
VIRAL MORPHOLOGY
They say we learn best by doing, so let's study
viral structure by making a virus, starting from the
nucleic acid inside and proceeding to the capsid and
envelope.
Nucleic Acid
Fig. 22-2. Viruses are classified as either DNA or
RNA viruses. So we have two choices for our virus: DNA
or RNA. Of course nothing is quite that simple. The nu-
cleic acid strands can be single-stranded, double-
stranded, linear, or looped, in separate segments or one
continuous strand. The nucleic acid sequences can en-
code a simple message or encode hundreds of enzymes
and structural proteins.
RNA Viruses
Let us choose RNA for the core of our virus. There are
3 types of RNA for our virus: positive (+) stranded, neg-
ative (-) stranded, or the RNA of the retroviruses.
The POSITIVE (+) means that the RNA is JUST
LIKE a messenger RNA (mRNA). When a positive (+)
stranded RNA virus enters a host cell, its RNA can im-
mediately be translated by the host's ribosomes into
protein.
Figure 22-2
When negative (-) stranded RNA viruses enter a cell,
they are not able to begin translation immediately.
They must first be transcribed into a positive (+) strand
of RNA (like mRNA). To do this, negative (-) stranded
RNA viruses must carry, in their capsid, an enzyme
called RNA-dependent RNA polymerase, which will
carry out the transcription of the negative (-) strand
into positive (+). Human cells do not have an RNA-de-
CHAPTER 22. VIRAL REPLICATION AND TAXONOMY
pendent RNA polymerase, so negative (-) stranded
viruses must carry their own.
Fig. 22-3.
Translation of positive (+) and negative (-)
RNA viruses.
One special RNA virus deserves mention here:
Retroviruses, of which the HN virus is a member.
Fig. 22-4.
The RNA of the retroviruses is transcribed
in a reverse fashion ("retrograde") into DNA! To do this,
these viruses carry a unique enzyme called reverse
transcriptase.
DNA Viruses
Fig. 22-5.
Unlike RNA, DNA cannot be translated di-
rectly into proteins. It must first be transcribed into
mRNA, with subsequent translation of the mRNA into
structural proteins and enzymes.
Every DNA virus has both a negative (-) strand and
a positive (+) strand. Here is the confusing part: The
positive (+) strand refers to the strand that is read,
while the negative (-) strand is ignored.
Fig. 22-6.
Unlike positive (+) stranded RNA, which is
translated directly into proteins, the positive (+)
stranded DNA is used as a template for transcription
into mRNA.
Figure 22-4
162
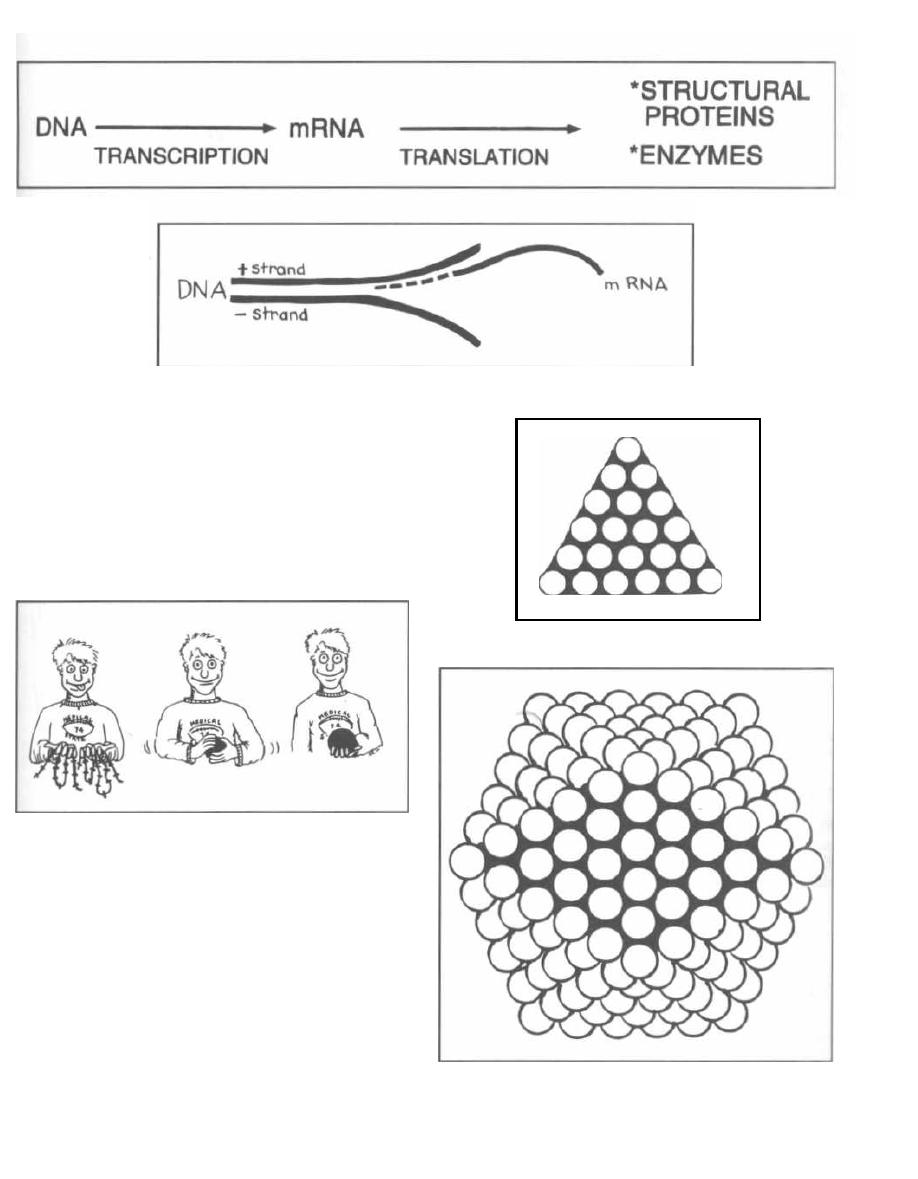
Figure 22-5
Figure 22-7
Fig. 22-7. Take 1 or more polypeptide chains and
organize them into a globular protein subunit. This will
be the building block of our structure and is called a
capsomer.
Fig. 22-5. Arrange the capsomers into an equilateral
triangle.
Fig. 22-9. Place 20 triangles together to form an
icosahedron.
Package the DNA or RNA inside the icosahedral
capsid!
Figure 22-6
Capsids
Now that we know about the different types of nucleic
acids, let's build a structure to house the genome. First
we will build a capsid. There are two types of capsids:
Icosahedral and helical.
Icosahedral Symmetry Capsids
CHAPTER 22.
VIRAL REPLICATION AND TAXONOMY
163
Figure 22-9
Figure 22-8
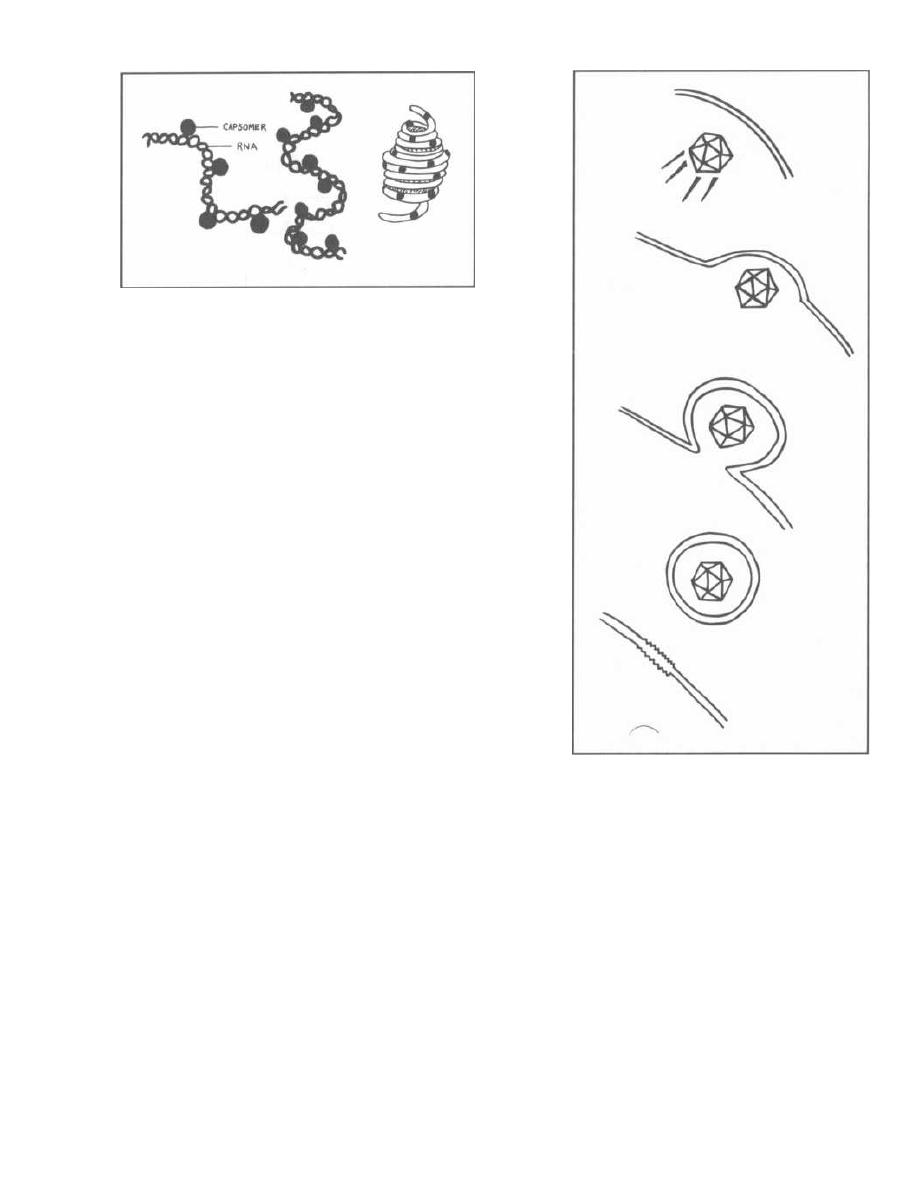
Figure 22-10
Helical Symmetry Capsids
Fig. 22-10. In helical symmetry the protein cap-
somers are bound to RNA (always RNA because only
RNA viruses have helical symmetry) and coiled into a
helical nucleoprotein capsid. Most of these assume a
spherical shape except for the rhabdoviruses (rabies
virus), which have a bullet-shaped capsid.
Envelope
Fig. 22-11. Now that we have made an icosahedral
capsid with a nucleic acid (RNA or DNA) inside and a
coiled helical nucleocapsid (RNA), let us cover the struc-
ture with a lipid bilayer membrane. Viruses acquire this
membrane by budding through the host cell nuclear or
cytoplasmic membrane and tearing off a piece of the
membrane as they leave There may be various glyco-
proteins embedded in their cell membranes.
Viruses that do not have membranes are referred to
as naked or nonenveloped. Those with membranes are
referred to as enveloped.
Finished Product
Fig. 22-12.
The appearance of the complete viruses
and their approximate sizes compared to the bacterium
E. coli,
and the very small bacteria,
Chlamydia, Rick-
ettsia,
and
Mycoplasma pneumoniae.
CLASSIFICATION
Viruses are classified according to their:
1) Nucleic acid:
Type of nucleic acid: DNA, RNA
Double- vs. single-stranded
Single or segmented pieces of nucleic acid
Positive (+) or negative (-) stranded RNA
Complexity of genome
CHAPTER 22.
VIRAL REPLICATION AND TAXONOMY
164
Figure 22-11
2) Capsid:
Icosahedral
Helical
3) Envelope:
Naked
Enveloped
4) Size:
The diameter of the helical capsid viruses
The number of capsomers in icosahedral capsids
We will now go over the virus families and the char-
acteristics that separate them.
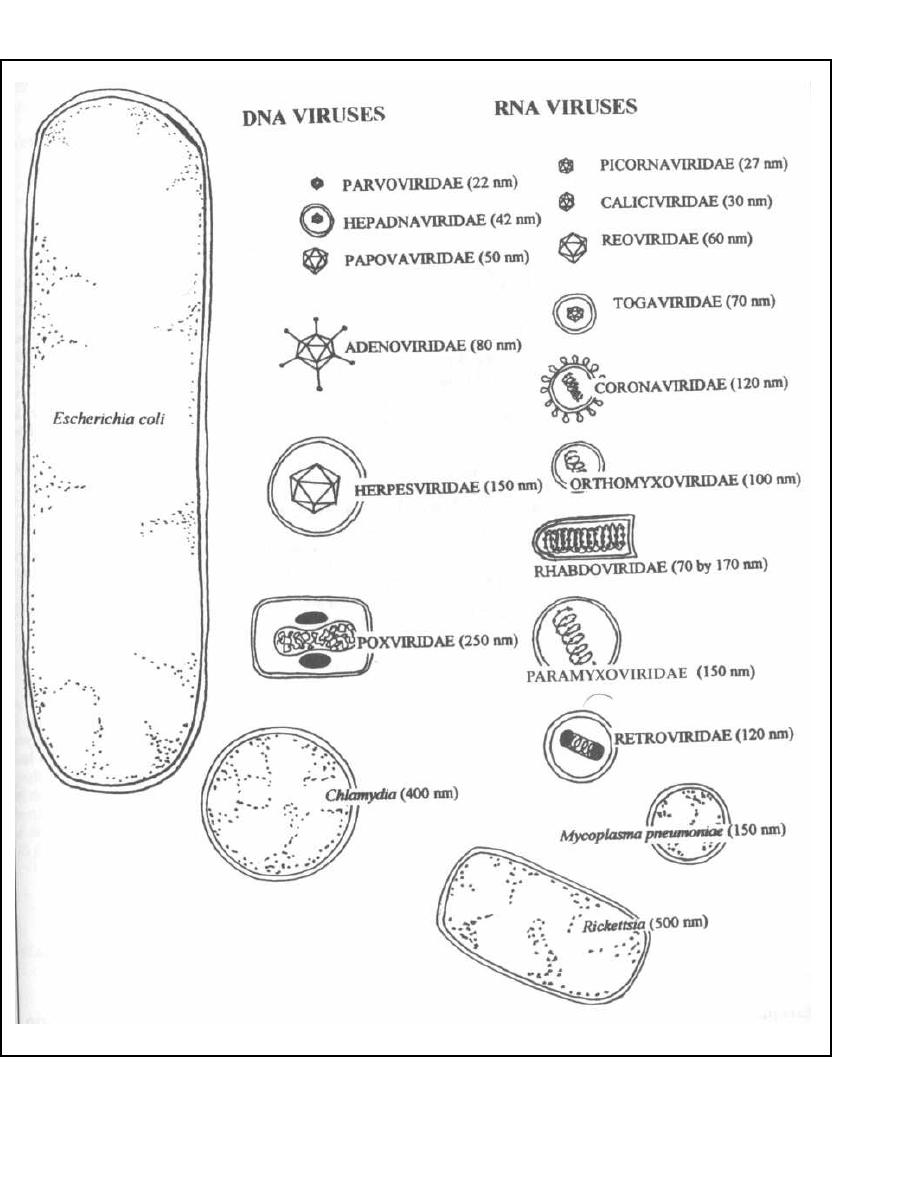
CHAPTER 22.
VIRAL REPLICATION AND TAXONOMY
Figure 22-12
165

DNA Viruses
These are sometimes referred to as the HHAPPPy
viruses:
Herpes
Hepadna
Adeno
Papova
Parvo
Pox
Most DNA viruses are double-stranded, show icosa-
hedral symmetry, and replicate in the nucleus (where
DNA customarily replicates).
Two DNA viruses break these rules:
1) Parvoviridae: This virus is so simple that it only
has a single strand of DNA. It is as simple as playing a
ONE PAR hole in golf.
2) Poxviridae: This virus is at the opposite end of
the spectrum and is extremely complex. Although it
does have double-stranded DNA, the DNA is complex in
nature, coding for hundreds of proteins. This virus does
not have icosahedral symmetry. The DNA is sur-
rounded by complex structural proteins looking much
like a box ( POX IN A BOX). This virus replicates in the
cytoplasm.
Three of the DNA viruses have envelopes:
Herpes Hepadna Pox
Three are naked: A woman must be naked for the
PAP smear exam.
PApova Adeno PArvo
Fig. 22-13.
The DNA viruses.
RNA Viruses
There are certain generalities about RNA viruses,
most of which are the opposite of DNA viruses.
Most RNA viruses are single-stranded (half are
positive [+1 stranded, half negative [-1), enveloped,
show helical capsid symmetry, and replicate in the
cytoplasm:
Exceptions:
1) Reoviridae are double-stranded.
2) Three are nonenveloped: Picorna, Calici, and
Reoviridae.
CHAPTER 22. VIRAL REPLICATION AND TAXONOMY
16 6
3) Five have icosahedral symmetry: Reo, Picorn
Toga, Flavi, Calici (Rhabdo has helical symmetry but
shaped like a bullet).
4) Two undergo replication in the nucleus: Retro
Orthomyxo.
Fig. 22-14. The RNA viruses.
VIRAL REPLICATION
Viruses cannot reproduce on their own. They must in-
vade a cell, take over the cell's internal machinery and
instruct the machinery to build enzymes and new viral
structural proteins. Then they copy the viral genetic
material enough times so that a copy can be placed in
each newly constructed virus. Finally, they leave the
host cell. The invading virus also blocks the synthesis of
any host DNA, RNA or proteins. This feature forces the
host cell to construct only viral proteins and copies of
the viral genetic material.
In order for viruses to reproduce, they must complete
these 4 steps:
1) Adsorption and penetration.
2) Uncoating of the virus.
3) Synthesis and assembly of viral products (as well
as inhibition of the host cell's own DNA, RNA and pro-
tein synthesis).
4) Release of virions from the host cell (either by ly-
sis or budding).
Adsorption and Penetration
Fig. 22-15.
The viral particle finds to the host cell
membrane. This is usually a specific interaction in
which a viral encoded protein on the capsid or a glyco
-
protein embedded in the virion envelope binds to a host
cell membrane receptor. Unlike the bacteriophage
virion (see Chapter 3 on Bacterial Genetics), which in-
jects its DNA, these viruses are completely internalized,
capsid and nucleic acid. This internalization occurs by
endocytosis or by fusion of the virion envelope with the
host cell membrane.
Transcription, Translation, Replication
RNA Viruses
These viruses usually undergo transcription, transla-
tion, and replication in the cytoplasm.
Toga
Orthomyxo
Corona
Paramyxo
Uncoating
Retro
Rhabdo
Picorna
Bunya
The nucleic acid is released from the capsid into the
Calici
Arena
nucleus or cytoplasm.
Reo
Fibo
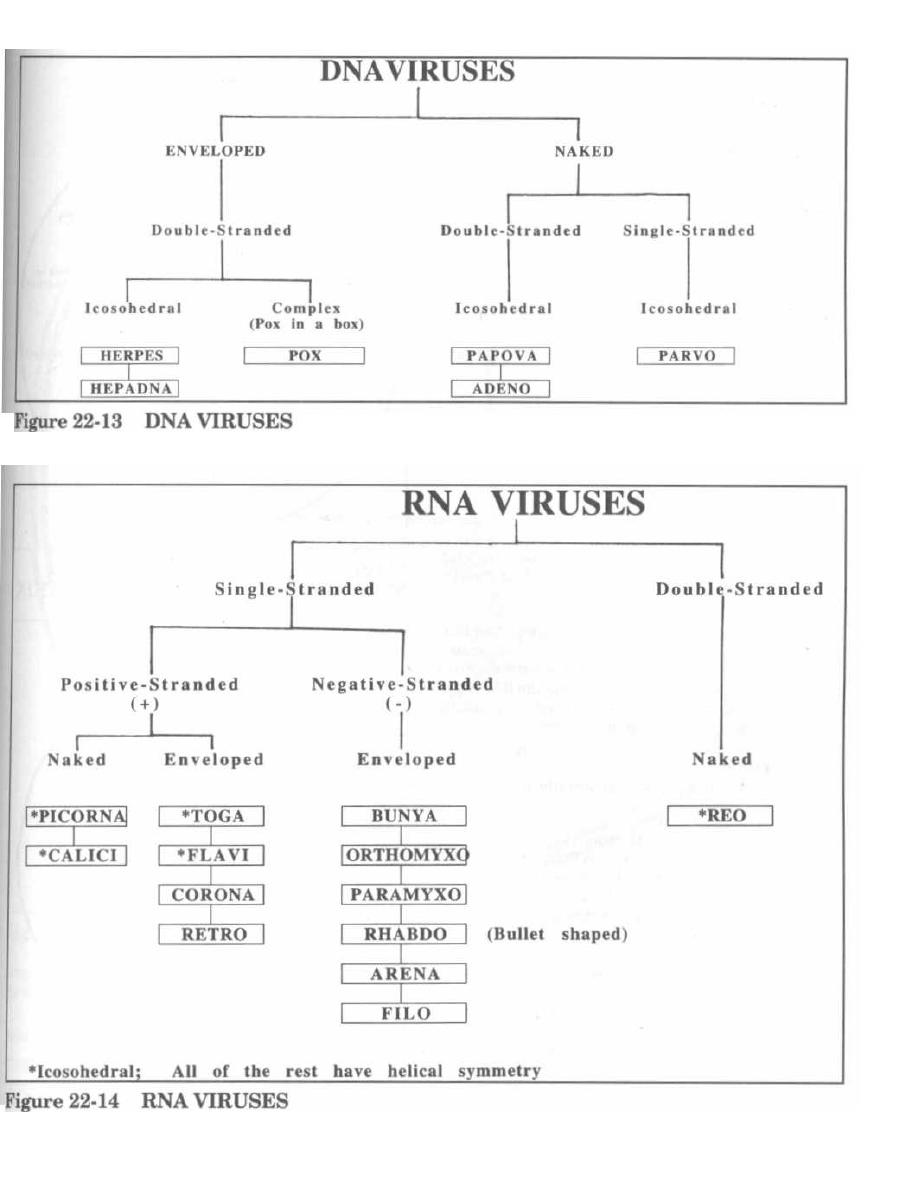
CHAPTER 22. VIRAL REPLICATION AND TAXONOMY
16 7
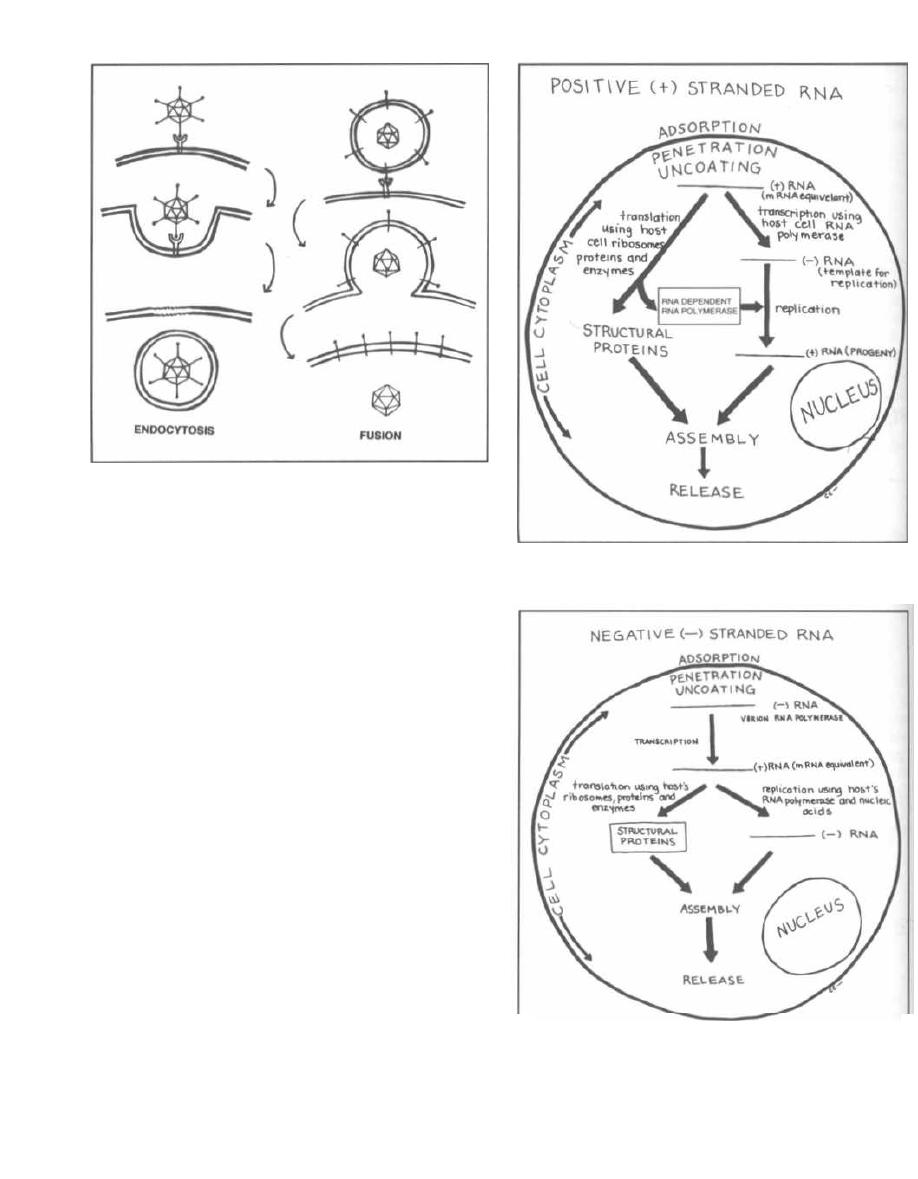
Figure 22-15
Fig. 22-16.
Positive (+) stranded RNA virus replica-
tion. These viruses do not carry an RNA dependent RNA
polymerase because they are read by the host directly
as mRNA.
Fig. 22-17
Negative (-) stranded RNA virus replica-
tion. The virus uncoats, releases a virion associated
RNA polymerase, and must first transcribe the negative
( -) strand to a positive (+) strand (using the RNA poly-
merase). The positive (+) strand then acts like mRNA
and undergoes both transcription and translation.
DNA
Viruses
Transcription and replication usually occur in the
nucleus.
Fig. 22-18.
DNA virus replication. DNA viruses tend
to be more genetically complex than RNA viruses. Thus,
viral transcription is divided into immediate early,
early, and late transcription. Another important con-
cept is that DNA viruses act in a similar fashion as our
own genome. Segments of DNA are transcribed in the
nucleus into mRNA, are spliced and processed, and the
mRNA then moves to the cytoplasm (endoplasmic retic-
ulum) where translation occurs.
Immediate early and Early: The initially tran-
scribed mRNA here encodes enzymes and proteins
needed for DNA replication and for further transcrip-
tion of late mRNA.
Late: The mRNA is usually transcribed after viral
DNA replication has begun and is transcribed from
CHAPTER 22. VIRAL REPLICATION AND TAXONOMY
16 8
Figure 22-16
POSITIVE (+)
STRANDED RNA VIRUS REPLICATION
Figure 22-17
NEGATIVE (- )
STRANDED RNA VIRUS REPLICATION
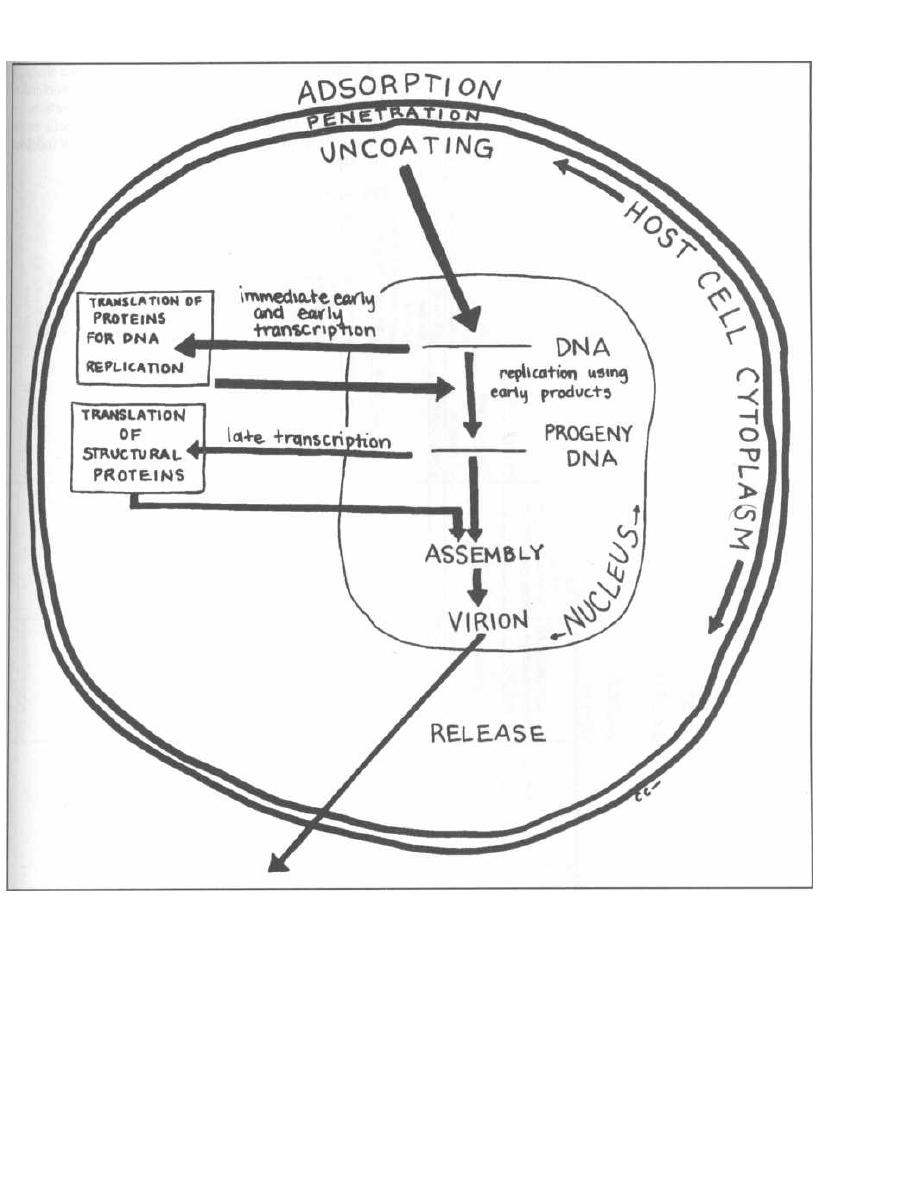
CHAPTER 22.
VIRAL REPLICATION AND TAXONOMY
Figure 22-18 DNA VIRUS REPLICATION
progeny DNA. The capsid structural proteins are syn-
thesized from the late mRNA genome.
Assembly and Release
The structural proteins and genome (RNA or DNA)
assemble into the intact helical or icosahedral virion.
The virion is then released.
169
Naked virions: The cell may lyse and release the
virions, or the virions may be released by reverse phago-
cytosis (exocytosis).
Enveloped virions: The newly formed naked virion
acquires its new "clothing" by budding through the
Golgi apparatus, nuclear membrane, or cytoplasmic
membrane, tearing off a piece of host cell lipid bilayer as
it exits.

HOST CELL OUTCOME
Death: With the viral infection, the host cell's own
function shuts down as the cell is commandeered for
virion replication. This can result in cell death.
Transformation: Infection can activate or introduce
oncogenes. This results in uncontrolled and uninhibited
cell growth.
CHAPTER 22. VIRAL REPLICATION AND TAXONOMY
17 0
Latent infection: The virus can survive in a sleeping
state, surviving but not producing clinically overt infec-
tion. Various factors can result in viral reactivation.
Chronic slow infection: Some viruses will cause
disease only after many years, often decades, of indolent
infection.
Fig. 22-19.
Summary of virus morphology.
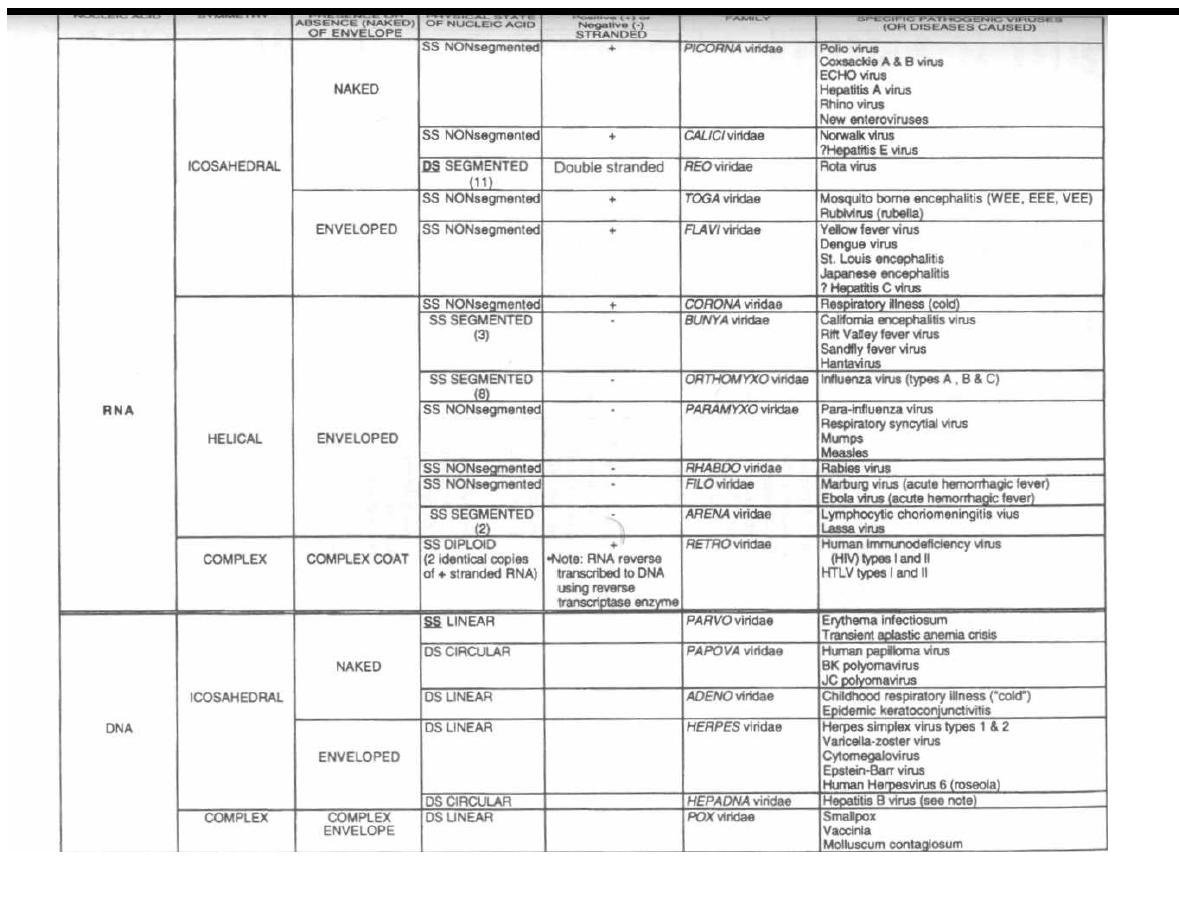
•Note: Deity virus (causes hepatitis) is an incomplete RNA virus. It needs the coinfection with of hepatitis B virus to cause disease
L'
O')1 Q
VIR Ai .
MORPHOLOGY
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
©MedMaster

CHAPTER 23. ORTHOMYXOVIRIDAE and PARAMYXOVIRIDAE
Figure 23-1
These 2 viral families have similar structures and the
ability to adsorb to glycoprotein receptors, particularly
in the upper respiratory tract. The orthomyxoviridae
are all influenza viruses, which cause the "ORdinary
flu." Paramyxoviridae also replicate in the upper respi-
ratory tract and can produce influenza-like illness, but
they also produce a PARAde of distinctly different dis-
eases. The paramyxoviridae include parainfluenza
virus, mumps, measles, and respiratory syncytial virus.
Fig. 23-1.
The orthomyxoviridae and paramyxoviridae.
ORTHOMYXOVIRIDAE
Understanding the structure of this virus will be im-
portant to you as a physician. The ability to produce epi-
demics and susceptibility to antibody immunity
d
vaccination all depend on the viral ultrastructure. You
will see at the end of this section that the paramyx-
oviridae have a similar structure with a few small
changes (making it oh so easy to learn!).
Fig. 23-2.
The orthomyxoviridae are spherical virions.
At the virion center lie 8 segments of negative ( -) stranded
RNA put together with a protein ( nucleocapsid protein
- NP) i nto a helical symmetry capsid. Surrounding the nu-
cleocapsid lies an outer membrane studded with long
glycoprotein spikes. There are 2 distinct types of glycopro-
tein: one with Hemagglutinin Activity (HA) and one
with Neuraminidase Activity (NA). Anchoring the
bases of each of these spikes on the inside of the viral lipid
bilayer are membrane proteins ( M-proteins).
Hemagglutinin (HA)
Fig. 23-3.
Hemagglutinin (HA) can attach to host
sialic acid receptors. Sialic acid receptors are present on
the surface of erythrocytes, so viruses with HA glyco-
proteins cause heme-agglutination when mixed with
red blood cells.
Host cell sialic acid receptors also exist on upper res-
piratory tract cell membranes, and HA binding to these
receptors activates fusion of the host cell membrane
with the virion membrane, resulting in dumping of the
172
Figure 23-2
SEGMENTED RNA
NUCLEOPROTEIN
(NP)
Figure 23-3
viral genome into the host cell. So HA is needed for ad
•
sorption! Antibodies against HA will block this binding
and prevent infection.
Neuraminidase ( NA)
Neuraminic acid is an important component of mucin,
the substance covering mucosal epithelial cells and
forming an integral part of the host's upper respiratory
defense barrier. As the name implies, neuraminidase
( NA) cleaves neuraminic acid and disrupts the mucin
barrier, exposing the sialic acid binding sites beneath.
1
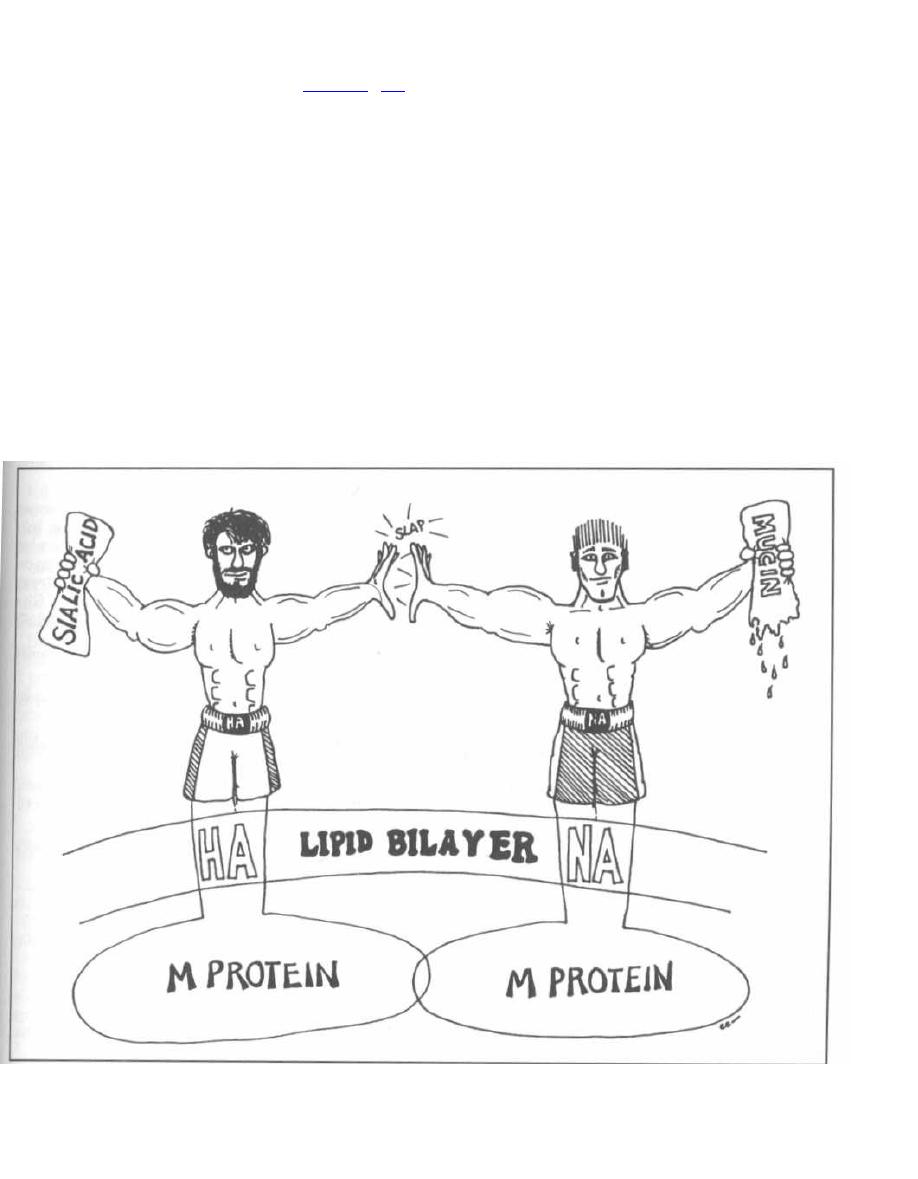
I
CHAPTER 23. ORTHOMYXOVIRIDAE AND PARAMYXOVIRIDAE
Fig. 23-4. Viral NA and HA act as tag team wrestling
partners, wrestling down the host's
cleaves the cell mucin barrier, while HA fuses to the
cell's sialic acid residues, enabling viral adsorption and
penetration.
Again, antibodies directed against NA are also
protective.
Influenza Serology and Epidemiology
There are 3 types of influenza virus: A, B, and C.
These types have many strains separated by antigenic
differences in HA and NA. Type A infects humans, other
mammals (swine, etc.), and birds. Type B and C have
only been isolated from humans.
When looking at the disease influenza, 2 questions
about epidemiology arise:
Q: If antibody to the NA and HA are protective, why
do we continually get epidemics of the bothersome flu,
with fever, chills, myalgias, arthralgias, headache, and
other miseries?
A: Antigenic Drift: During viral replication muta-
tions can occur in the HA or NA, leading to changes in
the antigenic nature of these glycoproteins. This is
termed antigenic drift because the changes are small,
just a little drift of the sailboat in the water. The re-
sulting new strains are only partially attacked by our
immune system, resulting in milder disease in adults
who have previously acquired antibodies. Major muta-
tional changes usually result in altered codon reading
frames and a nonviable virus.
Q: We all think that this is a pesky but mild self-lim-
iting disease. It can cause pneumonia and more serious
disease in the elderly, but usually it resolves without
complications in 3 to 7 days. So why have there been
devastating pandemics of influenza throughout his-
tory, as in 1918? Over a few weeks in 1918, 548,452 per-
sons in the U. S., 12.5 million persons in India, and 20
million persons worldwide died from this virus.
Figure 23-4
173

Figure 23-5
CHAPTER 23. ORTHOMYXOVIRIDAE AND PARAMYXOVIRIDAE
A: Antigenic Shift: Now we are really shifting
gears. We are taking the boat mentioned above and
airlifting it to a mountain in the Himalayas. With anti-
genic shift there is a complete change of the HA, NA,
or both. This can only occur with influenza type A
because the mechanism involves the trading of RNA
segments between animal and human strains. When 2
i nfluenza types co-infect the same cell, undergo replica-
tion and capsid packaging, RNA segments can be mis-
packaged into another virus. This virus now wields a
new HA or NA glycoprotein that has never been exposed
to a human immune system anywhere on the planet. So
the entire human population would be susceptible, lead-
ing to devastating pandemics.
The new HA and NA antigens are given number sub-
scripts to differentiate them. The pandemic of 1889
was caused by a virus with an H2 hemagglutinin, the
pandemic of 1900 was caused by a new virus with H3
hemagglutinin; in 1918 a swine flu virus transferred
its HA to a human virus and so was called Hswine
hemagglutinin (HSW). The chart below is only in-
cluded to demonstrate the many pandemics and their
new HA and NA antigenic composition.
Global pandemics:
1989:
H2N2
1900:
H3N2
1918:
H1N1: "Spanish flu"-highly pathogenic
strain-with high mortality (estimated
20-40 million deaths worldwide)
1947:
H1N1*
1957:
H2N2* "Asian flu"-illness but low mortality
1968:
H3N2* "Hong Kong flu"-illness but low
mortality
1977:
H1N1*
"Notice also that some strains caused a second pan-
demic as a new unexposed population grows to adulthood.
Fig. 23-5. Antigenic drift and shift: For influenza epi-
demics and pandemics to occur in people other than
children (who do not yet have antibodies), the antigens
in the NA and HA must somehow change, through
antigenic drift and antigenic shift.
174
In 1997, a new strain of avian influenza (Influenza A,
H5N1) was transmitted from infected poultry to humans
in Hong Kong. This avian flu virus contained super-
charged HA similar to that in the 1918 pandemic that
killed over 20 million people. The supercharged HA en-
abled the virus to kill almost half of the people who became
infected. Fortunately, the virus was poorly transmissible
to humans, and was controlled by destroying the poultry
in Hong Kong. This was certainly a close call.
Complications of Influenza
Even the normal yearly flu can cause complica-
tions. The elderly and immunocompromised suffer
more serious illness as the virus spreads to the lower
respiratory tract, resulting in pneumonia. The viral
infection also lowers the host defenses against
many bacteria. Secondary bacterial pneumonias by
Staphylococcus aureus, Streptococcus pneumoniae,
and others are common and the physician must follow
patients (especially the elderly) closely until complete
resolution of their illness. New fevers or failure to im-
prove means danger!!
Fig. 23-6. Study the figure of the child with the crown
(the Rey- Spanish for king) and the lightning bolts
around his head and liver. Children given aspirin when
they have influenza or varicella (chicken pox) can de-
velop a severe liver and brain disease called Reye's
Syndrome. It is not yet known why this occurs. Give
acetaminophen for fever in children, no aspirin!!!!
Diagnostic tests for influenza fall into
4
broad
categories:
1. Virus isolation: Culture of the virus allows for
genetic and antigenic analysis
2. Detection of viral proteins: New one hour tests help
guide the choice of antiviral agents
3. Detection of viral nucleic acid (RNA) in clinical
material is available by reverse transcription fol-
lowed by PCR (very sensitive method)
4. Serological diagnosis: 4-fold increase in specific
antibody levels over 2 weeks

Figure 23-6
Treatment and Control
Influenza viruses are grown in mass quantities in
chick embryos, which are then inactivated, purified, and
used as vaccines. Scientists carefully choose 3 strains
that are circulating in the population or expected to cause
an outbreak in the next season. These vaccines have vari-
able success depending on the accuracy of the "guess-
work." The vaccines should be given to the elderly,
immunocompromised patients, and health-care workers.
There are now a few choices for treating the flu.
Amantadine and rimantidine prevent the uncoating
of influenza A. They can both prevent influenza A
infection and can reduce the severity of symptoms.
Sanamavir (inhaled) and oseltamivir (oral) are neu-
raminidase (NA) inhibitors, which can shorten the
course of influenza A and B.
PARAMYXOVIRIDAE
The structure of paramyxoviridae is very similar to
that of the orthomyxoviridae. The differences are that:
1) The negative (-) stranded RNA is in a single
strand, not segmented.
2) HA and NA are a part of the same glycoprotein
spike, not 2 different spikes.
3) They possess a fusion (F) protein (not present in
the orthomyxoviridae) that causes the infected host
cells to fuse together into multinucleated giant cells
(syncytial cells similar to those caused by herpesviridae
and retroviridae infection).
There are 4 paramyxoviridae that cause human dis-
ease: parainfluenza virus, respiratory syncytial virus,
CHAPTER 23. ORTHOMYXOVIRIDAE AND PARAMYXOVIRIDAE
1
175
mumps virus, and measles virus. Before reading about
each, let's examine the big picture:
1) Think lungs: All adsorb to and replicate in the
upper respiratory tract. Respiratory syncytial virus and
parainfluenza virus both cause lower respiratory infec-
tions (pneumonia) in children and upper respiratory
tract infections (bad colds) in adults.
2) Think kids: Most infections occur in children.
3) Think viremia: The viral infection results in dis-
semination of virions in the blood to distant sites.
Mumps and measles reproduce in the upper respiratory
tract and spread hematogenously to distant organs.
Mumps can produce local parotid and testes infection
(parotitis and orchitis), and measles can produce a se-
vere systemic febrile illness. Brain infection (encephali-
tis) can occur with both mumps and measles.
Parainfluenza Virus
The parainfluenza virus causes upper respiratory in-
fection in adults ranging from cold symptoms such as
rhinitis, pharyngitis, and sinus congestion, to bronchi-
tis and flu-like illness. Children, elderly, and the im-
munocompromised also suffer from lower respiratory
tract infections (pneumonia).
Croup is a parainfluenza infection of the larynx and
other upper respiratory structures (laryngotracheo-
bronchitis) that occurs in children. Swelling of these
structures produces airway narrowing. Stridor (a
wheezing sound) and a barking cough (like a seal) occur
as air moves through the narrowed upper airways.
Respiratory Syncytial Virus (RSV)
RSV is so-named because it causes respiratory infec-
tions and contains an F-protein that causes formation of
multinucleated giant cells (syncytial cells). This virus
differs from the rest of its kin by lacking both the HA
and NA glycoproteins.
RSV is the number one cause of pneumonia in young
children, especially in infants less than 6 months of age.
The virus is highly contagious with outbreaks occurring
in winter and spring. The treatment of RSV infection
is less than ideal, with ribavirin studies showing con-
flicting results. Efforts have therefore focused on pre-
vention. RSV infection can be prevented in a high
percentage of cases with palivizumab, which is a
monoclonal antibody against RSV that is produced by a
recombinant DNA method. A blood-derived product,
serum RSV immune globulin, is also available, but
comes with the risk of transmission of blood-borne
infections.
Previously infected persons are not entirely immune,
but the subsequent infections are usually limited to the
upper respiratory tract.
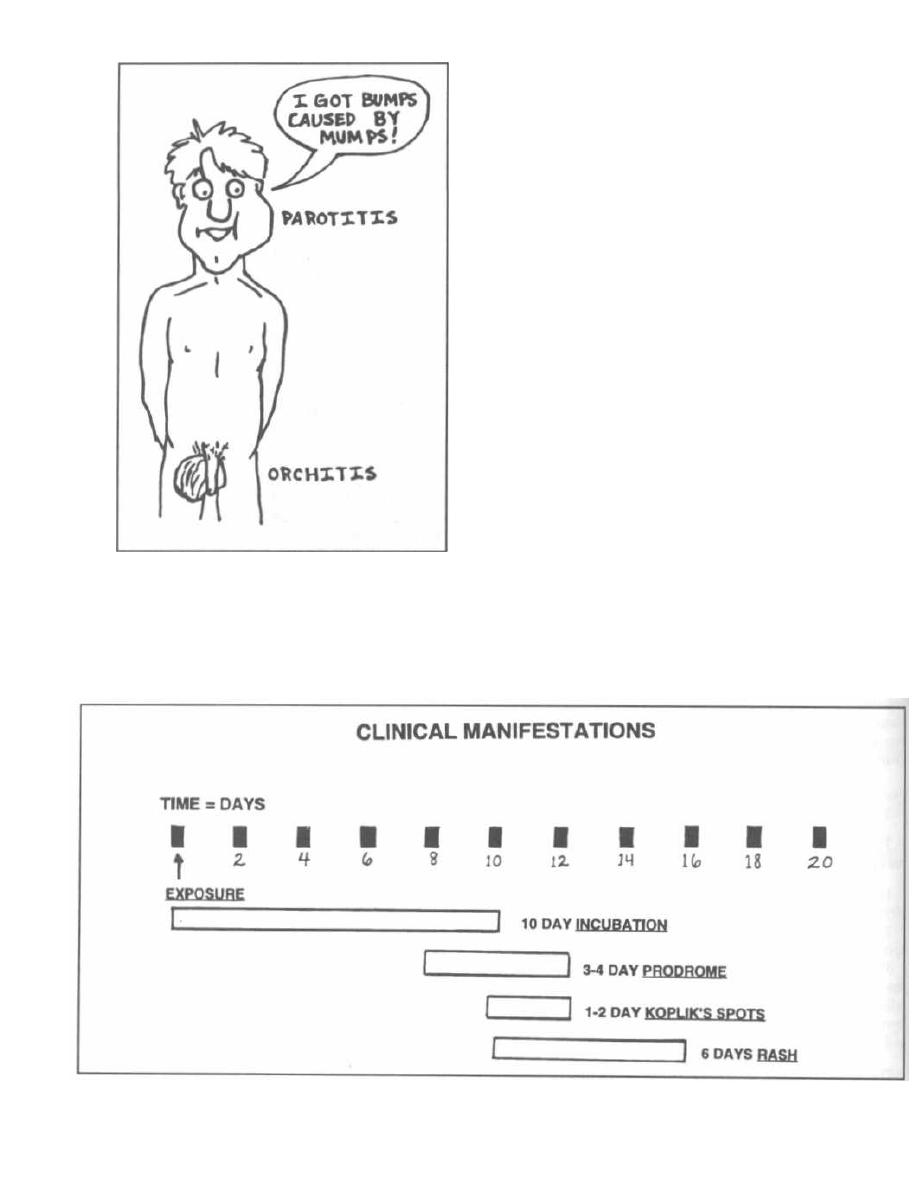
Figure 23-7
Figure 23-8
CHAPTER 23. ORTHOMYXOVIRIDAE AND PARAMYXOVIRIDAE
176
Mumps Virus
Mumps virus replicates in the upper respiratory tract
and in regional lymph nodes and spreads via the blood
to distant organs. Infection can occur in many organs,
but the most frequently involved is the parotid gland.
Fig. 23-7. About 3 weeks after initial exposure to
mumps virus the parotid gland swells and becomes
painful. The testes are also frequently infected. About
25% of infected males who have reached puberty can de
velop orchitis. The testes enlarge and stretch the cap-
sule, resulting in intense pain. Infertility is a rare
complication. Meningitis and encephalitis can also oc-
cur, the former being more common and less severe.
There is only one antigenic type, and a live attenu-
ated viral vaccine is a part of the trivalent measles-
mumps-rubella (MMR) vaccine.
Measles Virus
Due to the effectiveness of the MMR vaccine, there
were only 2,900 cases of measles in the U. S. in 1988.
However, the disease continues to have worldwide im-
pact with about 2 million deaths annually.
Fig. 23-8. The clinical manifestations of measles (also
called rubeola).
Exposure
Measles
\
virus is highly contagious and spreads
through nasopharyngeal secretions by air or by

CHAPTER 23. ORTHOMYXOVIRIDAE AND PARAMYXOVIRIDAE
Figure 23-9
Figure 23-10
direct contact. The virus multiplies in the respira-
tory mucous membranes and in the conjunctival
membranes. Incubation lasts for 2 weeks prior to the
development of rash.
Prodrome
Fig. 23-9. Measles prodrome. Prior to the appearance
of the rash, the patient suffers from prodromal illness
with conjunctivitis, swelling of the eyelids, photophobia,
high fevers to 105° F, hacking cough, rhinitis, and
malaise (feels cruddy).
177
Koplik's Spots
Fig. 23-10. Koplik's spots. A day or 2 before the rash,
the patient develops small red-based lesions with blue-
white centers in the mouth. Think of a cop licking a red-
white-blue lollipop.
Rash
The measles rash is red, flat to slightly bumpy (mac-
ulopapular). It spreads out from the forehead to the face,
neck, and torso, and hits the feet by the third day.

CHAPTER 23. ORTHOMYXOVIRIDAE AND PARAMYXOVIRIDAE
Figure 23-11
Fig. 23-11.
As the measles rash spreads downward,
the initial rash on the head and shoulders coalesces.
The rash disappears in the same sequence as it devel-
oped. Visualize a can of measles-brand red paint be-
ing poured over a patient's head. The paint is thicker
over the head and shoulders and drips completely off
in 6 days.
Complications
Like mumps, the measles virus disseminates to many
organ systems and can damage those sites, causing pneu-
monia, eye damage, heart involvement (myocarditis), and
the most feared complication, encephalitis. Encephalitis
is rare, but 10% of patients who develop this will die.
Infection with measles during pregnancy does not
cause birth defects but has been associated with spon-
taneous abortion and premature delivery. In fact,
measles in pregnant women results in fetal death in
20% of cases.
178
Subacute sclerosing panencephalitis (SSPE) is a
slow form of encephalitis caused by measles virus. Many
years after a measles infection the child or adolescent
may have slowly progressing central nervous system
disease, with mental deterioration and incoordination.
The MMR vaccine, which contains live attenuated
measles virus, is preventative.
Fig. 23-12. Summary of the orthomyxoviridae and
paramyxoviridae.
Recommended Review Articles:
Cox NJ, Subbarao K Influenza. The Lancet
1999;354:1277-1281.
Domachowske J, Rosenberg H. Respiratory Syncytial Virus
Infection: Immune Response, Immunopathogenesis, and
Treatment. Clinical Microbiology Reviews April 10, 1999;
298-309.
Steinhauer D. Role of Hemagglutinin Cleavage for the Patho-
genicity of Influenza Virus. Virology
1999; 258: 1-20.
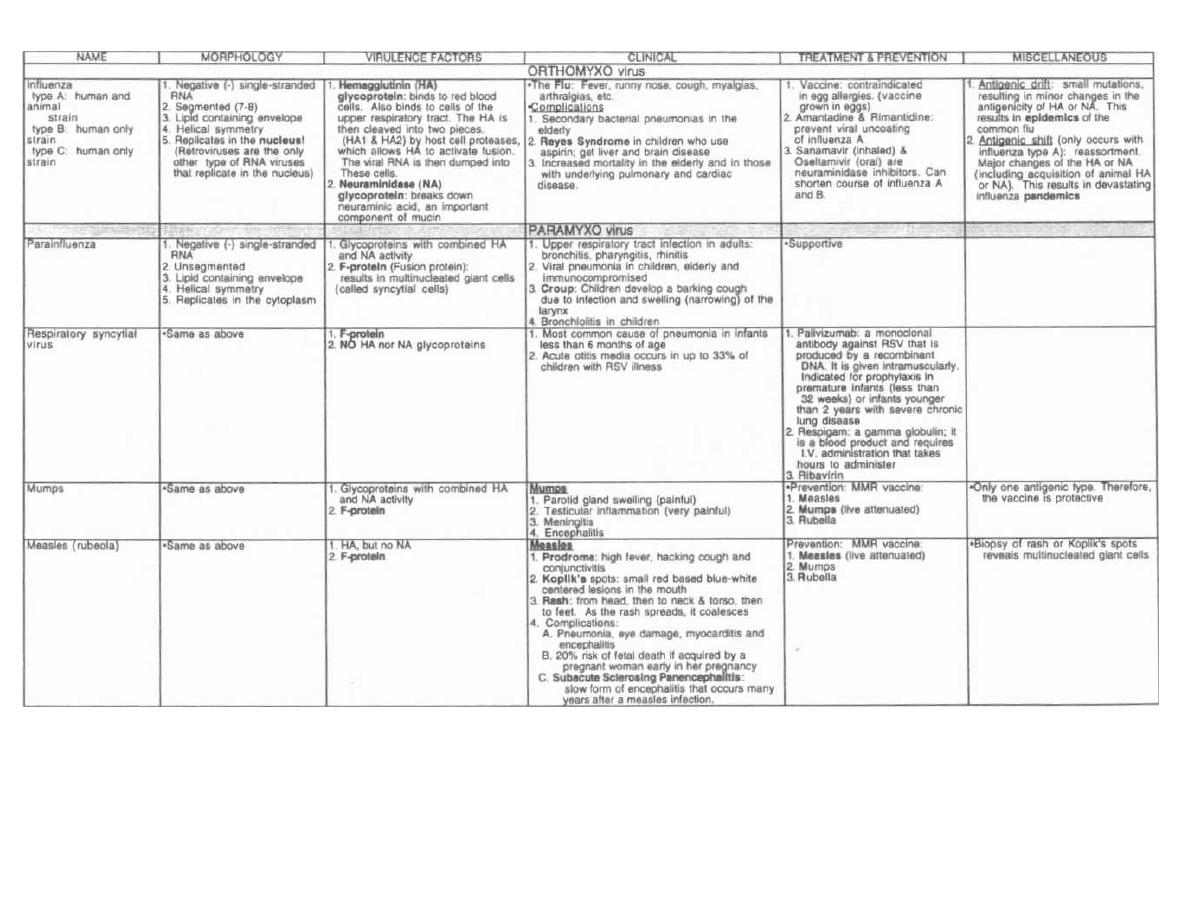
Figure 23-12
ORTHOMYXOVIRIDAE AND PARAMYXOVIRIDAE
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
OMedMaster

CHAPTER 24. HEPATITIS VIRIDAE
Viral hepatitis is an infection of the liver hepatocytes by
viruses. There are 5 known viruses that infect the liver.
The 5 RNA viruses are:
1) Hepatitis A virus (HAV).
2) Hepatitis C virus (HCV), which was previously
called NON-A NON-B until it was isolated.
3) Hepatitis D virus (HDV).
4) Hepatitis E virus (HEV).
5) Hepatitis G virus.
There is 1 DNA virus called:
1) Hepatitis B virus (HBV)
Hepatitis A and E are both transmitted via the fecal-
oral route, while the rest are transmitted via blood-to-
blood (parenteral) contact. Just as A and E are at both
ends of ABCDE, so they are transmitted by elements of
both ends of the GI tract. A = Anal, E = Enteric,
BCD = BlooD.
We will now discuss the clinical disease hepatitis and
then cover each virus in more detail.
VIRAL HEPATITIS
Viral hepatitis can be a sudden illness with a mild to
severe course followed by complete resolution. This is
called acute viral hepatitis and can be caused by all
of these viruses. Hepatitis can also have a prolonged
course of active disease or silent asymptomatic infection
termed chronic viral hepatitis. The parenterally
(blood-to-blood) transmitted HBV, HCV, and HDV can
cause chronic hepatitis.
1) Acute viral hepatitis has a variable incubation
period, depending on the virus type. The growth of the
virus first results in systemic symptoms much like the
flu, with fatigue, low-grade fever, muscle/joint aches,
cough, runny nose, and pharyngitis. One to two weeks
later the patient may develop jaundice as the level of
bilirubin, which is normally cleared by the liver, rises.
As the virus grows in the hepatocytes, these liver cells
necrose (die). The hepatocytes produce enzymes that are
released during cell death. These are the liver-function
enzymes aspartate aminotransferase (AST), alanine
aminotransferase (ALT), gamma-glutamyl transpepti-
dase (GGT), and alkaline phosphatase. Elevated blood
levels of these liver enzymes help establish the diagno-
sis of hepatitis.
So about 2 weeks into the illness, the patient is often
jaundiced, has a painful enlarged liver and high blood
levels of liver-function enzymes.
180
The details of how to determine which virus is caus-
ing the hepatitis will be discussed with each virus.
2) Chronic viral hepatitis is more difficult to diag-
nose because the patient is often asymptomatic with
only an enlarged tender liver and mildly elevated liver.
function enzyme levels.
The Pattern of Liver Enzyme Elevation
Different diseases result in different patterns of liver.
function enzyme elevation. For example, viral hepatitis
usually causes the transaminases ALT and AST to ele-
vate to very high levels, while GGT, alkaline phos-
phatase, and bilirubin are only mildly elevated. As the
disease progresses, bilirubin levels rise higher. A gall-
stone in the bile duct causes the opposite to occur: biliru-
bin, alkaline phosphatase, and GGT rise higher than
ALT and AST. The reason for this is
a fellows.
Fig. 24-1.
The hepatocytes produce AST and ALT.
The cells that line the bile canaliculi produce alka-
line phosphatase and GGT. The bile canaliculi carry
bilirubin.
Fig. 24-2.
With viral hepatitis there is cell necrosis as
the virus takes over the cell machinery. The hepatocytes
rupture, resulting in the release of AST and ALT. Some
pericanalicular cells will also be destroyed. So we will
see very high AST and ALT with a little elevation of al-
kaline phosphatase and GGT. As the infection worsens,
the liver swells and the canaliculi narrow, resulting in
a backup of bilirubin into the blood. So with viral he-
patitis the ALT and AST are very high, and the biliru-
bin, alkaline phosphatase, and GGT rise later in the
course.
Fig. 24-3.
A stone blocking the bile duct would re-
sult in:
1) Inability to excrete the bilirubin, resulting in high
blood levels of bilirubin.
2) The backup results in increased alkaline phos-
phatase and GGT synthesis by the canalicular cells.
Imagine the backup of pressure popping the cells, re-
sulting in the release of alkaline phosphatase and GGT
(not the true mechanism but a good memory tool).
So with an obstructive process (stone in bile duct), AST
and ALT would only be minimally elevated; alkaline
phosphatase, GGT, and bilirubin would be very elevated.
Hepatitis A Virus (HAV)
HAV has a naked icosahedral capsid with a posi-
tive (+) single-stranded RNA nucleic acid. It is in the
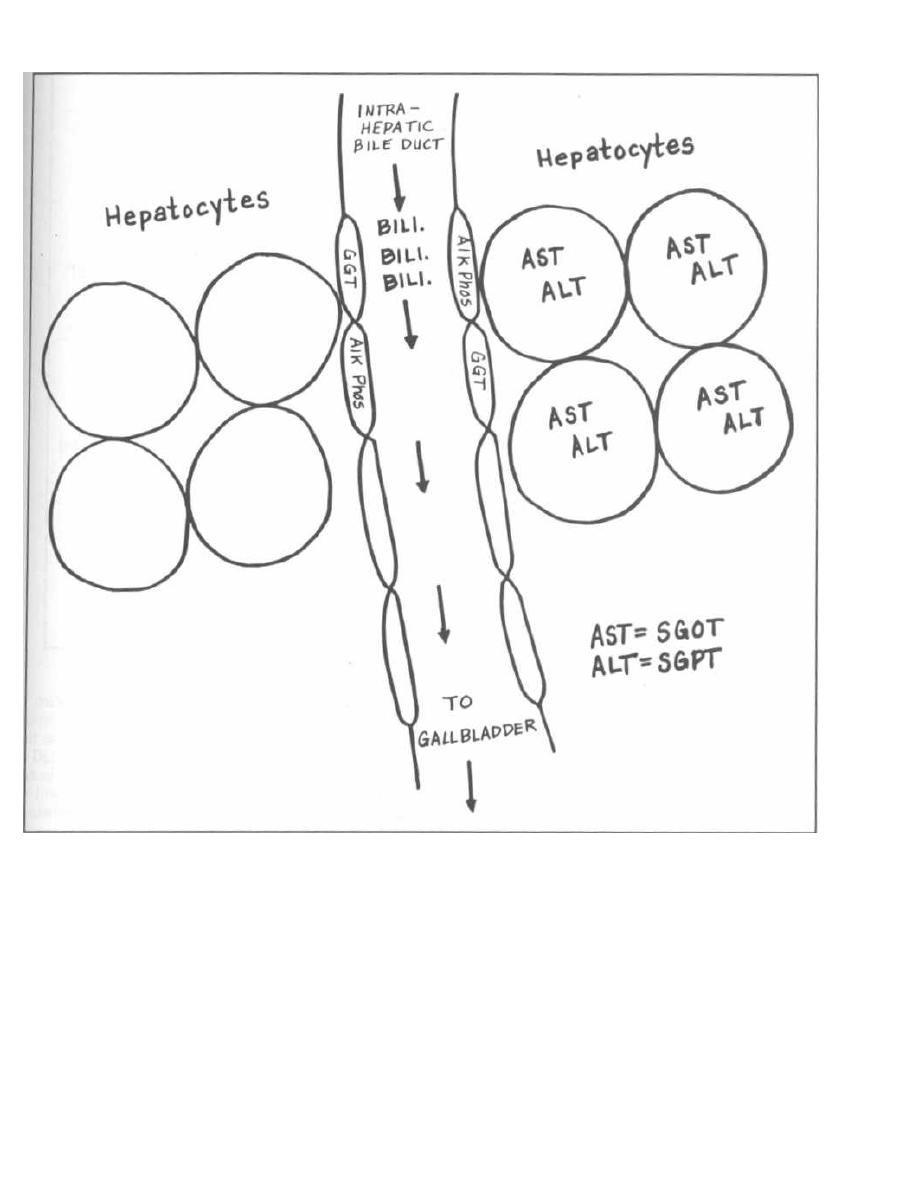
Figure 24-1
family Picornaviridae, and as is the case with most of
this family it is transmitted by the fecal-to-oral route
(HAV = Anus).
Epidemiology
HAVe you washed your hands??? About 25,000 cases
of hepatitis A infection are reported each year in the
U.S., and there are many more infections that are
asymptomatic or unreported. In fact, 40% of Americans
living in urban centers have serologic evidence of prior
infection, but only 5% remember the infection. Out-
CHAPTER 24. HEPATITIS VIRIDAE
181
breaks often occur secondary to fecal-to-hand-to-mouth
contact. Examples of this include an infected food han-
dler contaminating food after poor hand washing, per-
sons ingesting fecally contaminated drinking water, or
close person-to-person contact in institutions such as
day care centers. There is a 15-40 day incubation period
(about 1 month) before the patient develops acute he-
patitis as described earlier.
Young children are the most frequently infected, and
they have a milder course than do adults, often without
developing jaundice or even symptoms. At the other end
of the spectrum, a small percentage (1-4%), usually
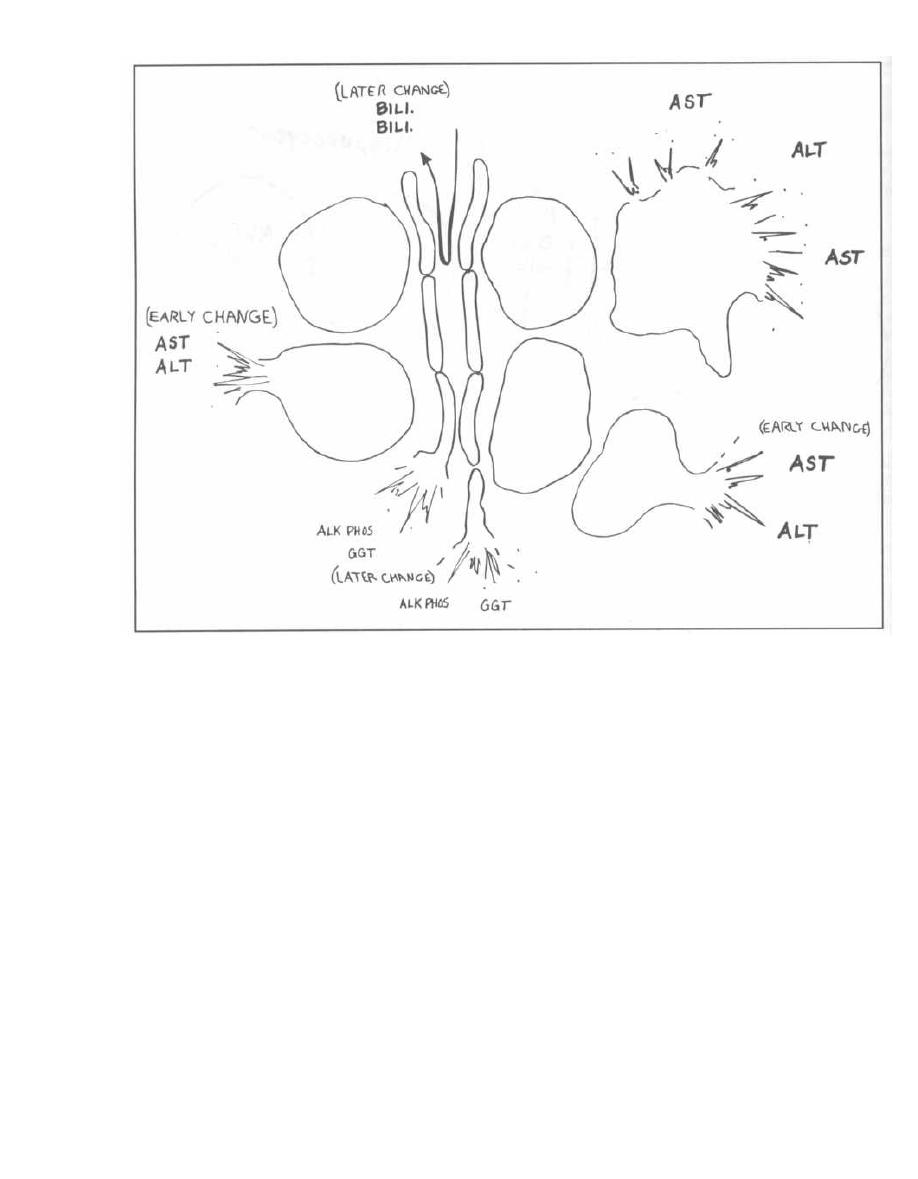
Figure 24-2
adults, will develop fulminant (severe) hepatitis. How-
ever, death from HAV is very rare (1%).
Serology
Serologic tests can help establish the diagnosis. The
HAV capsid is antigenic, resulting in the host produc-
tion of anti-HAV IgM and later, the anti-HAV IgG. A pa-
tient with active infection will have anti-HAV IgM
detectable in the serum. Anti-HAV IgG indicates old in-
fection and no active disease. This antibody lasts indef-
initely and is protective, which means that it will
protect against future infection with HAV.
Fig. 24-4. Hepatitis A infection: A time line of clinical
symptoms and antibody development.
Fig. 24-5.
Hepatitis A serology.
Treatment
A new vaccine is available and may be given to peo-
ple at high risk of HAV infection, such as travelers. If a
person has been exposed, pooled immune serum globu-
CHAPTER 24. HEPATITIS VIRIDAE
182
lin will prevent or decrease the severity of infection, if
given early during incubation. Pooled immune serum
globulin is obtained by ethanol fractionation from the
plasma of hundreds of donors. Since anti-HAV IgG is
present in about 40% of the population, there will be an-
tibody in the pooled immune serum globulin that will in-
activate the virus. Once infection is established,
treatment is only supportive.
Hepatitis B Virus (HBV)
You will have an intimate relationship with HBV
throughout your career. Why is this? In an infected pa-
tient, this virus lives in all human body fluids (semen,
urine, saliva, blood, breast milk. .. ). As a physician,
you will come into contact with patients who harbor this
virus. Since hospital workers are considered to be an at-
risk group, you will receive immunization against HBV.
So you will touch this virus frequently and will actually
harbor antibodies against it.
HBV = Big and Bad
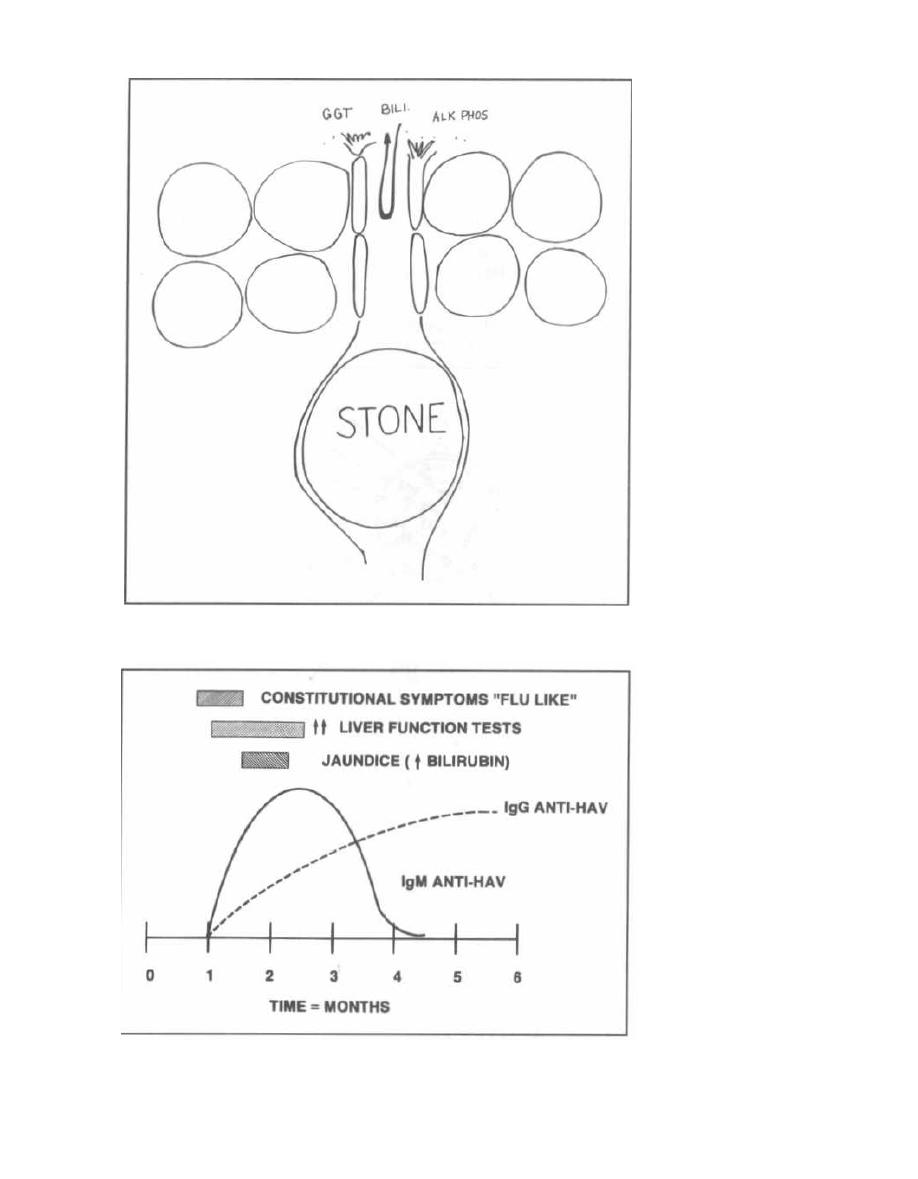
Figure 24-3
CHAPTER 24. HEPATITIS VIRIDAE
Figure 24-4
183
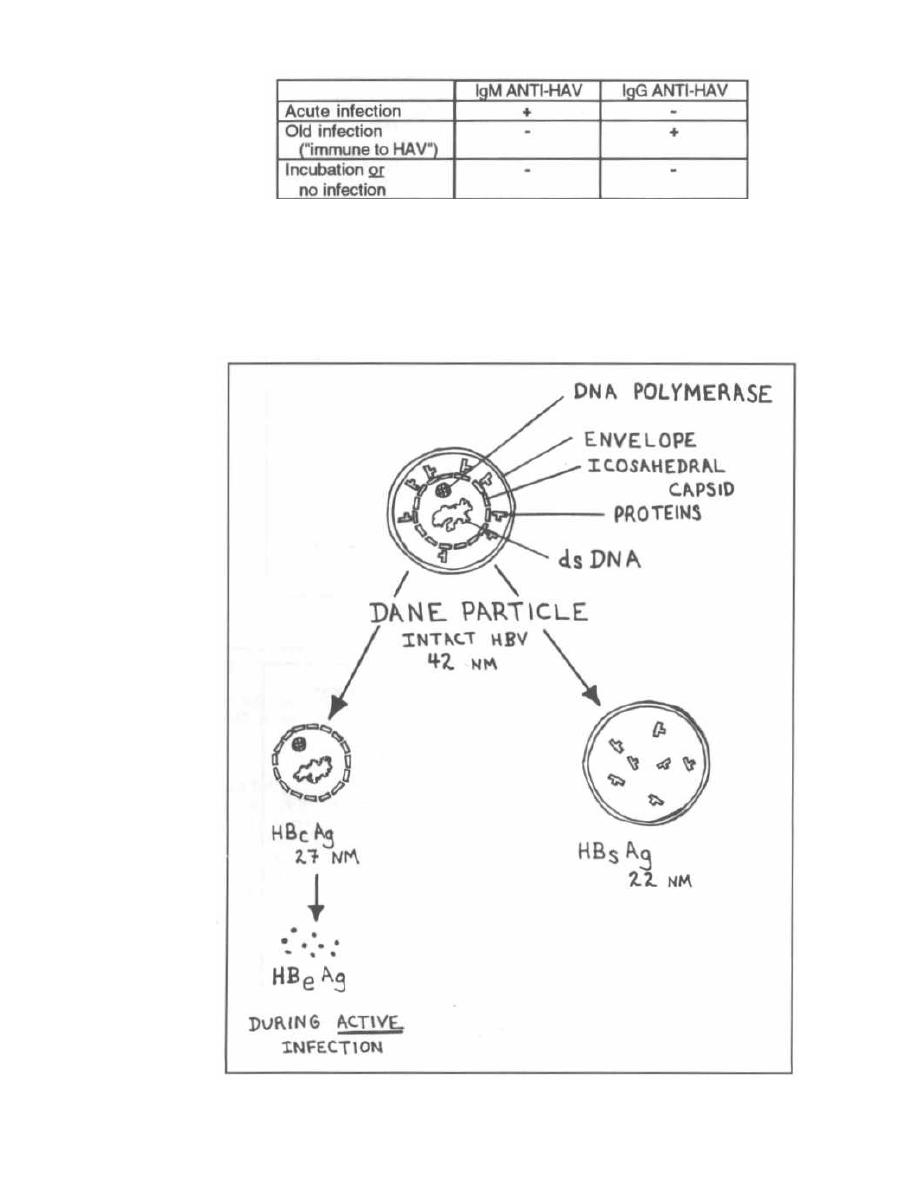
HBV is very different from HAV. It is a Big (42 NM)
virus with an enveloped icosahedral capsid and double-
stranded circular DNA.
Fig. 24-6. The intact virus is called the Dane parti-
cle ( Big like a Great Dane) and looks like a sphere un-
der electron microscopy. Notice that the Dane particle
has an envelope and an icosahedral capsid studded with
protein spikes. In its core is a double-stranded DNA
with associated DNA polymerase enzyme.
When looking at infected blood with electron mi-
croscopy, you will notice the Dane particle spheres, de
•
CHAPTER 24. HEPATITIS VIRIDAE
Figure 24-6
184
Figure 24-5
SEROLOGY OF HEPATITIS A

scribed above, as well as longer filamentous structures.
These filamentous structures (as seen under electron
microscopy) are composed of the envelope and some cap-
sid proteins that have disassociated from the intact
virion. This part of the virus is called the hepatitis
B surface antigen (HBsAg) and is of critical impor-
tance because antibodies against this component (anti-
HBsAg) are protective. Having anti-HBsAg means the
patient is immune against HBV.
Removing HBsAg leaves the viral core, which is
called hepatitis B core antigen (HBcAg) and is also
antigenic. However, antibodies against the core (anti-
HBcAg) are not protective (do not result in immunity).
During active infection and viral growth, a soluble
component of the core is released. This is called HBeAg.
This antigen is a cleavage product of the viral core struc-
tural polypeptide. HBeAg is found dissolved in the
serum, and is a marker for active disease and a
highly infectious state. Pregnant mothers with
HBeAg in their blood will almost always transmit HBV
to their offspring (90% transmission rate), whereas
mothers who have no HBeAg will rarely infect the
neonate (10% transmission rate).
Epidemiology
HBV is present in human body fluids and is transmit-
ted from blood-to-blood contact. This non-oral transmis-
sion is called parenteral transmission. Transmission
from an infected patient can occur by needle sharing, ac-
cidental medical exposures (needle sticks, blood spray,
touching blood with unprotected hands), sexual contact,
blood transfusions, perinatal transmission, etc. This
virus is extremely contagious.
Pathogenesis
Another reason that HBV is a Bad dude is that
unlike hepatitis A, which can only cause an acute
hepatitis, HBV can cause acute and chronic hepati-
tis. The following are disease states caused by BIG
BAD HBV:
1) Acute hepatitis.
2) Fulminant hepatitis: Severe acute hepatitis
with rapid destruction of the liver.
3) Chronic hepatitis:
a) Asymptomatic carrier: The carrier patient
never develops antibodies against HBsAg (anti-
HBsAg) and harbors the virus without liver injury.
There are an estimated 200 million carriers of HBV
in the world.
b) Chronic-persistent hepatitis: The patient
has a low-grade "smoldering" hepatitis.
CHAPTER 24. HEPATITIS VIRIDAE
185
c) Chronic active hepatitis: The patient has an
acute hepatitis state that continues without the nor-
mal recovery (lasts longer than 6-12 months).
4) Co-infection
with
hepatitis
delta
virus
(HDV): See HDV section.
Liver injury appears to occur from a cell-mediated im-
mune system attack on HBV. Viral antigens on the sur-
face of infected hepatocytes are targets for cytotoxic
T-cells. Immune complexes of antibody and HBsAg can
deposit in tissues and activate the immune system, re-
sulting in arthritis, as well as skin and kidney damage.
Patients who have immunosuppressed states, such as
malnutrition, AIDS, and chronic illness, are more likely
to be asymptomatic carriers because their immune sys-
tem does not attack.
Complications
Primary hepatocellular carcinoma is a compli-
cation of HBV. With chronic infection the HBV DNA
becomes incorporated into the hepatocyte DNA and
triggers malignant growth. There is a 200X increase in
the risk of developing primary hepatocellular carcinoma
in HBV carriers as compared to noncarriers.
Infection with HBV can result in permanent liver scar-
ring and loss of hepatocytes. This is called cirrhosis.
Serology
Serologic tests help establish HBV infection. The
many antigens and antibodies are simpler than they
seem, as follows:
1) HBsAg: The presence of HBsAg always means
there is LIVE virus and infection, either acute, chronic,
or carrier. When anti-HBsAg develops, HBsAg disap-
pears and the patient is protected and immune.
a) HBsAg = DISEASE (chronic or acute)
b) Anti-HBsAg = IMMUNE, CURE, NO AC-
TIVE DISEASE!!!
2) HBcAg: Antibodies to HBcAg are not protective
but we can use them to understand how long the infec-
tion has been ongoing. With acute illness we will see
IgM anti-HBcAg. With chronic or resolving infection
IgG anti-HBcAg will develop.
a) IgM anti-HBcAg = NEW INFECTION
b) IgG anti-HBcAg = OLD INFECTION
3) HBeAg: The presence of HBeAg connotes a high
infectivity and active disease. Presence of anti-HBeAg
suggests lower infectivity.
a) HBeAg = HIGH INFECTIVITY, virus go-
ing wild!
b) anti-HBeAg = LOW INFECTIVITY
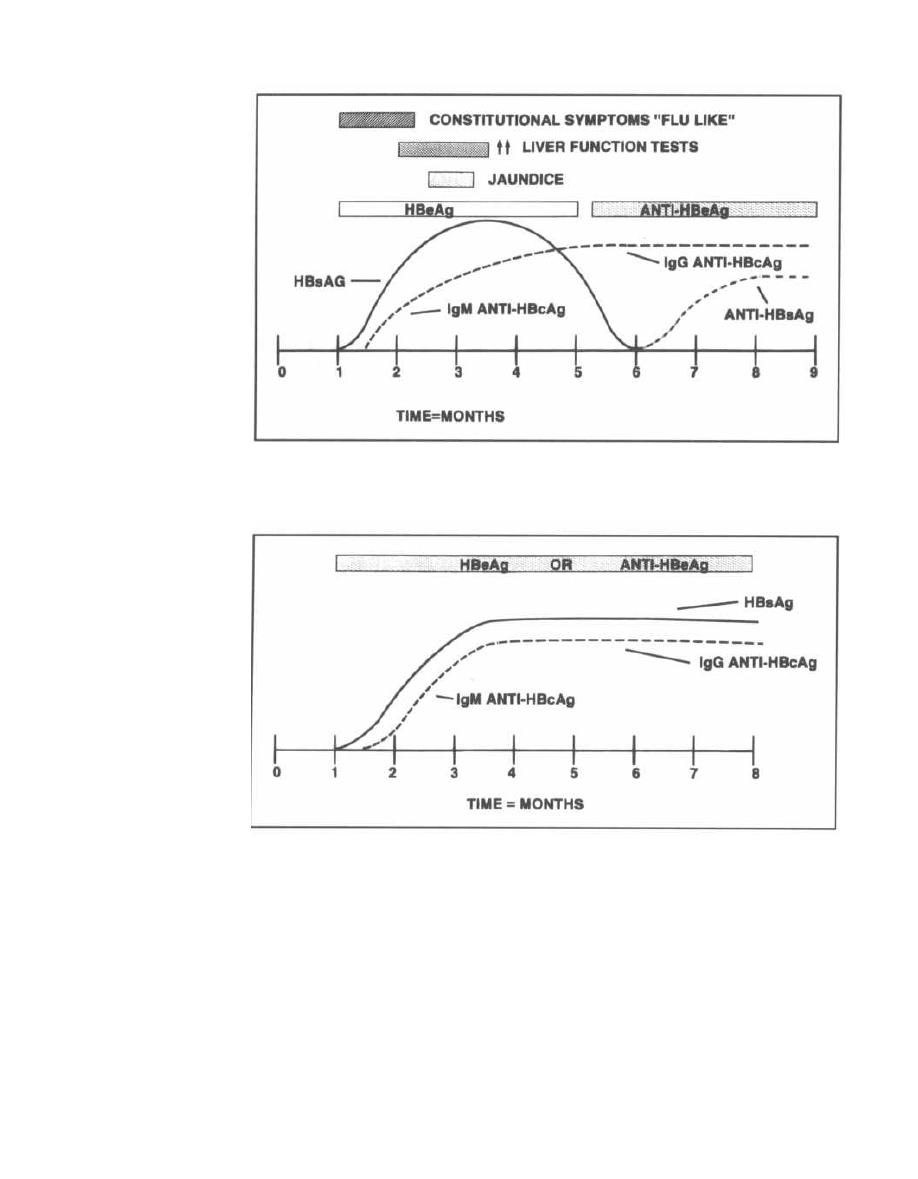
Figure 24-7
Fig. 24-7.
Time course of acute HBV infection, with
complete resolution and immunity.
Figure 24-8
Fig. 24-8.
Time course of chronic HBV infection, with
failure to develop the protective anti-HBsAg antibodies.
Treatment
Prevention and control of hepatitis B involves:
1) Serologic tests on donor blood to remove HBV-
contaminated blood from the donor pool.
2) Active immunization: The vaccine is a recombinant
vaccine. The gene coding for HBsAg is cloned in yeast and
used to produce mass quantities of HBsAg, used as the
CHAPTER 24. HEPATITIS VIRIDAE
18 6
vaccine. There is no risk of developing disease from the
vaccine because it contains only the surface envelope and
proteins (HBsAg = no DNA or capsid). The HBV vaccine
is now given to all infants at birth, 2, 4, and 15 months; it
is also given as 3 injections to adolescents and high-risk
adults (health care workers, IV drug users, etc.).
3) Anti-viral agents for treatment of chronic active or
persistent HBV infection:
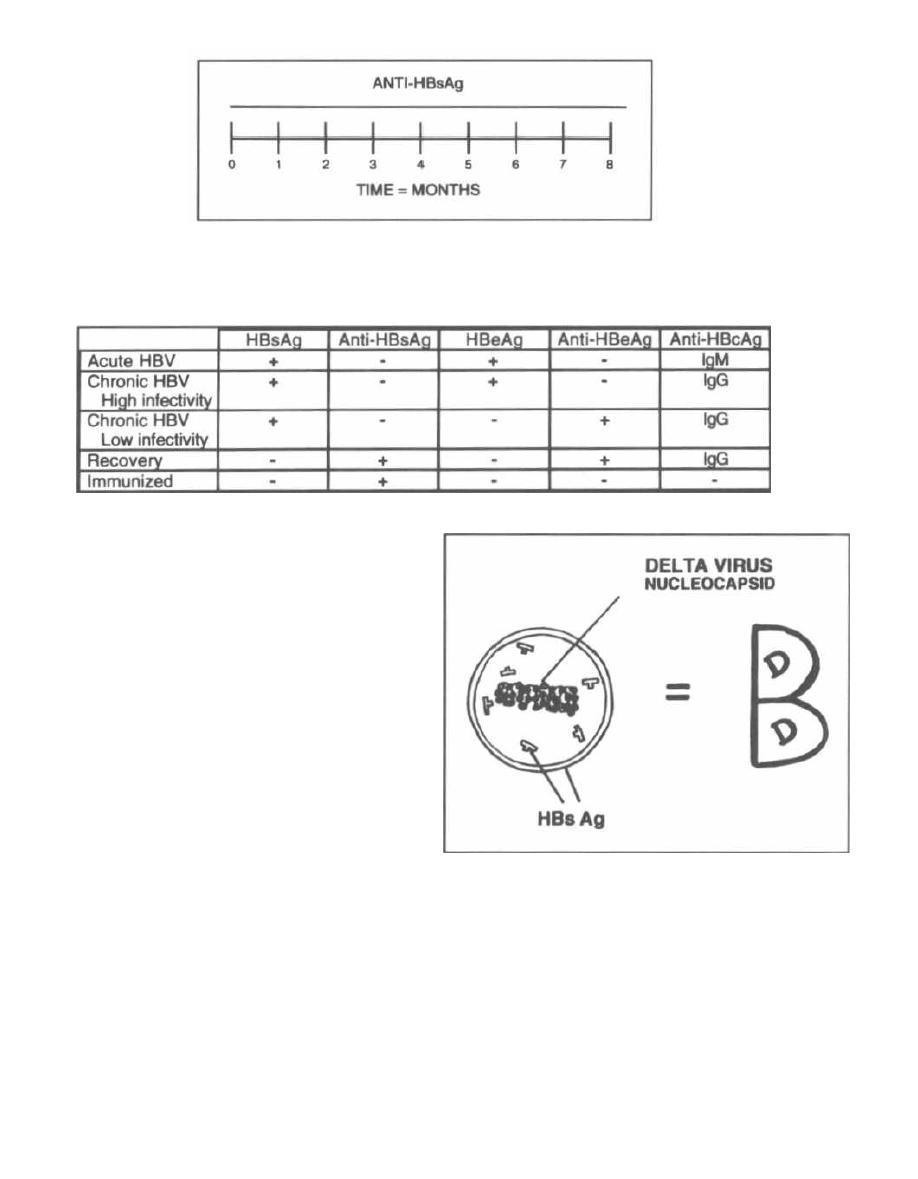
Figure 24-9
Fig. 24-9.
Serology of the medical student vaccinated
with HBV
recombinant vaccine.
Figure 24-10 SEROLOGY OF HEPATITIS B
Fig. 24-10.
Hepatitis B serology.
a) The anti-HIV drug lamivudine suppresses
HBV DNA to undetectable levels, but most patients
have relapses when the drug is discontinued. (Dien-
stag, 1995).
b) Treatment with interferon alpha suppressed
HBV DNA and HBeAg in 50% of treated patients
and appears to prevent cirrhosis in these respon-
ders (Niederau, 1996). New trials of combination
therapy with interferon alpha and ribavirin are
planned for patients with chronic active hepatitis
B or C.
Hepatitis Delta Virus (HDV)
This RNA virus is transmitted parenterally and
can only replicate with the help of HBV. The delta
virus helical nucleocapsid actually uses HBV's enve-
lope, HBsAg. HDV steals the clothes from HBV and can
only cause infection with the HBsAg coat.
Fig. 24-11.
Notice the HBV envelope and proteins
surround the HDV helical nucleocapsid. Next to it is a
conceptual figure of the letter D in a big B.
HBV+ HDV= Big Bad Dude
Without Big Bad HBV, HDV is just a dud and is not
infectious. Infection occurs in 2 ways:
CHAPTER 24. HEPATITIS VIRIDAE
187
Figure 24-11
1) Co-infection: HBV and HDV both are transmit-
ted together parenterally (IV drug use, blood transfu-
sions, sexual contact, etc.) and cause an acute hepatitis
similar to that caused by HBV. Antibodies to
HBsAg will be protective against both, ending the
infection.
2) Superinfection: HDV infects a person who has
chronic HBV infection (like the 200 million worldwide
HBV carriers). This results in acute hepatitis in a

patient already chronically infected with HBV. This
HDV infection is often severe, with a higher incidence of
fulminant hepatitis, cirrhosis, and a greater mortality
(5-15%). The patient with chronic HBV cannot make
Anti-HBsAg and so remains chronically infected with
both HBV and HDV.
Serology is currently not very helpful for diagnostic
purposes because IgM and IgG anti-HDV are in the
serum for only a short period. As there is no treatment,
control of HBV infection is currently the only way to pro-
tect against HDV.
Hepatitis C Virus (HCV)
Blood products were first screened for the presence of
HBV, and the incidence of post-transfusion hepatitis de-
clined. However, there was still a risk of developing non-
A, non-B hepatitis. Recently an etiologic agent has been
identified and called hepatitis C virus (HCV). Most of
the cases (about 90%) of non-A, non-B hepatitis are
caused by HCV. Blood products are now also screened
for HCV.
HCV is transmitted parenterally and has been iden-
tified as an enveloped icosahedral RNA virus. It causes
acute and chronic hepatitis in a similar manner as HBV.
In fact, HCV should be called hepatitis Cirrhosis
virus because half of those infected develop chronic he-
patitis. A large percentage of these develop chronic ac-
tive hepatitis and eventual cirrhosis. Hepatocellular
carcinoma can develop in patients with chronic active
HCV infection and cirrhosis.
Anti-HCV antibodies develop months after exposure
and are used to screen donor blood products and aid in
the diagnosis of HCV induced hepatitis.
Persons who develop chronic active HCV infection
have a high likelihood of developing cirrhosis and liver
failure. For patients with chronic active HCV, treat-
ment with alpha-interferon can sometimes result in res-
CHAPTER 24. HEPATITIS VIRIDAE
olution of liver inflammation (about 50% of treated pa-
tients will respond).
The problem is that treatment with interferon alpha
makes patients feel like they have the flu, and only
10-25% will have lasting remissions after therapy is
discontinued (Poynard, 1995).
Treatment with the anti-viral drug ribavirin only
temporarily normalized liver enzymes in
1
/3
of patients
(Di Bisceglie, 1995).
Hepatitis E Virus (HEV)
HEV hepatitis is often referred to as non-A hepati-
tis because it shares similarities with HAV. HEV is also
transferred by the fecal-oral route, frequently with the
consumption of fecally contaminated water during mon-
soon flooding. The E stands for Enteric (fecal-oral). Itis
endemic to Asia, India, Africa, and Central America.
Hepatitis G Virus
Hepatitis G is an RNA virus in the
Flavivirus
family.
It is transmissible by transfusion & parenteral routes.
It has not conclusively shown to cause liver disease.
Fig. 24-12.
Summary of hepatitis viruses.
References
Dienstag JL, et al. A preliminary trial of lamivudine for
chronic hepatitis B infection. N Engl J Med 1995;333:
1657-61.
Di Bisceglie AM, et al. Ribavirin as therapy for chronic active
hepatitis C: a randomized, double-blind, placebo-controlled
trial. Ann Int Med 1995;123:897-903.
Niederau C, et al. Long-term follow-up of HBeAg-positive pa-
tients treated with interferon alpha for chronic hepatitis B.
N Engl J Med 1996;334:1422-7.
Poynard T, Bedossa P, et al. A comparison of three inter-
feron alpha-2b regimens for the long term treatment
of chronic non-A, non-B hepatitis. N Engl J Med 1995;
332:1457-62.
Figure 24-12 HEPATITIS VIRIDAE
M. Gladwin and B. Trattler,
Clinical
Microbiology Made Ridiculously Simple
©MedMaster

Figure 25-1
The retroviridae are a large group of RNA viruses that
i nfect animals and humans.
Fig. 25-1.
There are 3 big concepts unique to the
retroviridae: RETRO, GROW, and BLOW.
1)
Retro: Most RNA viruses (negative- or positive-
stranded), enter the host cell and act as mRNA or are
transcribed into mRNA. The retroviridae are different.
They carry with them a unique enzyme called reverse
transcriptase. This enzyme is an RNA-dependent
DNA polymerase that converts the viral RNA into
DNA. This viral DNA has unique "sticky" ends that
allow it to integrate into the host's own DNA, as
do transposons.
2) Grow: Retroviruses can cause cancer in the cells
they infect. Genes called oncogenes i n humans and an-
i mals can cause the malignant transformation of nor-
mal cells into cancer cells. Some retroviruses carry
oncogenes in their genome. Inactive oncogenes in ani-
mals and humans are called proto-oncogenes. These
are genetic time bombs waiting for activation, which
can occur by carcinogen-induced DNA mutation or by
retrovirus infection.
3) Blow: Some retroviridae are cytotoxic to certain
cells, blowing them up. The most notable is the human
i mmunodeficiency virus (HIV) which destroys the T-
helper lymphocytes it infects. This ultimately results in
devastating immunodeficiency.
In this chapter we will discuss: 1) oncogenes; 2) the
human retroviridae, using HIV as a model for retroviri-
dae structure and genetics; and 3) HIV infection and
4) the Acquired Immunodeficiency Syndrome (AIDS)
caused by HIV.
CHAPTER 25.
RETROVIRIDAE, HIV, and AIDS
ONCOGENES
190
What Is Cancer?
Normal cells have strict growth control. They divide
an exact number of times with exact timing, depending
where in the body they are located. When normal cells
are grown on a plate of nutrient media, they form a
single layer and stop dividing when they touch each
other. This is called contact inhibition. Malignant
cells lose contact inhibition and pile up in vitro. They
become immortal, dividing continuously as long as
there is nutrient supply. In the body this uncontrolled
growth causes physical damage by local expansion,
steals nutrients from other cells, produces abnormally
high levels of hormones or other cell products, and can
i nfiltrate areas where other cells grow (such as the
bone marrow).
How Do Oncogenes Cause Cancer?
The normal cell has receptor proteins in the cell mem-
brane that regulate cell growth. Growth factors (mito.
gems) bind to these receptors to regulate growth.
Examples of these receptors are the insulin receptor,
the Epidermal Growth Factor (EGF) receptor, and the
Platelet Derived Growth Factor (PDGF) receptor. Mito-
gen stimulation of these receptors causes intracellular
phosphorylation of tyrosine. Phosphotyrosine then acts
as an intracellular growth messenger.
Fig. 25-2.
The Rous sarcoma virus oncogene (src), en-
codes a transmembrane protein that also phosphory-
lates tyrosine, but at ten times the normal rate!!!
The erb-B oncogene that causes cancer in chickens en-
codes a protein similar to the EGF receptor. Other onco-
genes encode proteins that are similar to the PDGF and
i nsulin receptors!
Still other oncogenes encode proteins that act at
the nuclear level to promote uncontrolled expression of
growth genes.
Retroviridae and Oncogenes
Because most of the retroviridae isolated to date
cause either leukemia or sarcoma in their vertebrate
hosts, they are sometimes called leukemia sarcoma
viruses.
Some retroviruses cause cancer directly
( acute) by integrating an intact oncogene into the host
DNA. Others cause cancer indirectly (nonacute) by acti-
vating a host proto-oncogene.
Figure 25-2
Acute Transforming Viruses
Fig. 25-3.
Some retroviridae, the acute transforming
viruses, carry intact oncogenes within their viral
genome, which when integrated into the host DNA
cause malignant transformation. This integration is
facilitated by the "sticky" ends and an enzyme called
integrase.
The acute transforming viruses were discovered in
1911 when Peyton Rous injected cell-free filtered mate-
rial from a chicken tumor into another chicken. The
chicken subsequently developed tumor. The causative
agent is now known to be a retrovirus called the Rous
sarcoma virus which possesses within its DNA an in-
tact oncogene called src. When the Rous sarcoma virus
infects a cell, it reverse transcribes its RNA into DNA.
The DNA is integrated into the host's genome by inte-
grase. Once integrated the src gene is expressed, caus-
ing malignant transformation.
Where Do Viral Oncogenes Come From?
Studies have shown that normal host DNA has se-
quences that are homologous to viral oncogenes but are
still inactive (proto-oncogenes). These genes most likely
are involved in cell growth regulation. Somehow, during
normal viral infection and integration, a mistake in
splicing occurs and a virus "captures" a proto-oncogene.
This proto-oncogene ultimately becomes activated in
the virus so the virus now carries an oncogene.
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
191
The oncogene gene sequence is so long that most
acute transforming viruses have lost their own RNA
critical for viral replication. These viruses are called de-
fective acute transforming viruses and require a co-
infecting virus to cause cancer.
The Rous sarcoma virus is the only known acute
transforming virus that is non-defective. It has the full
RNA genome needed for replication and also carries an
accessory src oncogene.
Non-acute Transforming Viruses
Fig. 25-4.
Other retroviridae, the non-acute trans-
forming viruses, activate host cell proto-oncogenes by
i ntegrating viral DNA into a key regulatory area. These
viruses do not carry oncogenes and thus have room for
the full genome necessary for viral replication.
HUMAN RETROVIRUSES
By the mid-1970's, retroviruses had been discovered
in many vertebrate species, including apes. The hy-
pothesis that humans may also be infected with retro-
viruses led to a search that ultimately resulted in the
isolation of a retrovirus from the cell lines and blood of
patients with adult T-cell leukemia. This virus is called
human T-cell leukemia virus (HTLV-I).
HTLV-I has now been linked to a paralytic disease
that occurs in the tropics (Caribbean islands) called
t ropical spastic paraparesis. HTLV-1 induced
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
Figure 25-3
leukemia has also been described in the Caribbean
and Japan.
A second human retrovirus was isolated from T-cells
of patients with a T-cell variant of hairy cell leukemia.
This is called HTLV-II, but this virus has no known role
in producing disease.
In early 1980 a new epidemic was first noted that we
now call the Acquired Immunodeficiency Syn-
drome (AIDS). Several factors suggested that this dis-
ease was caused by a retrovirus:
1) The infectious agent was present in filtered blood
products, such as concentrated factor VIII given to
patients with hemophilia. This suggested a viral etiol-
ogy, as something small would be necessary to pass
the filters.
2) There was a delayed onset between exposure (sex-
ual or blood products) and the development of disease.
This delayed onset had been observed in the other
known retroviral diseases.
3) Immunodeficiency occurs with other animal retro-
viruses, such as feline leukemia virus. Even HTLV-I can
cause immunosuppression.
4) AIDS patients have destruction of the T-helper
lymphocytes. The known human retroviruses HTLV-I
and II were both T-cell tropic.
192
Investigators stimulated T-cell culture growth (T
-
cells from patients infected with AIDS) with inter-
leukin-2 and were able to find RNA and DNA,
suggesting a retroviral etiology. The virus, which
was subsequently identified, was called the human
i mmunodeficiency virus (lily).
A second retrovirus, called HIV-2, causes a dis
-
ease similar to AIDS in western Africa. It is a dis
-
tantly related virus with 40% sequence homology with
HIV-1.
A virus that causes an AIDS-like disease in primates,
simian immunodeficiency virus (SIV), shares a close se-
quence homology with HIV-2.
HN STRUCTURE
The structure of the virion and genome is similar for
all of these retroviruses. We will focus on HIV, the cause
of the world's most feared current epidemic. HIV is at
the center of intensive research aimed at halting its dev-
astating disease, AIDS.
Fig. 25-5.
Under the electron microscope, HIV ap-
pears as a spherical enveloped virion with a central
cylindrical nucleocapsid.
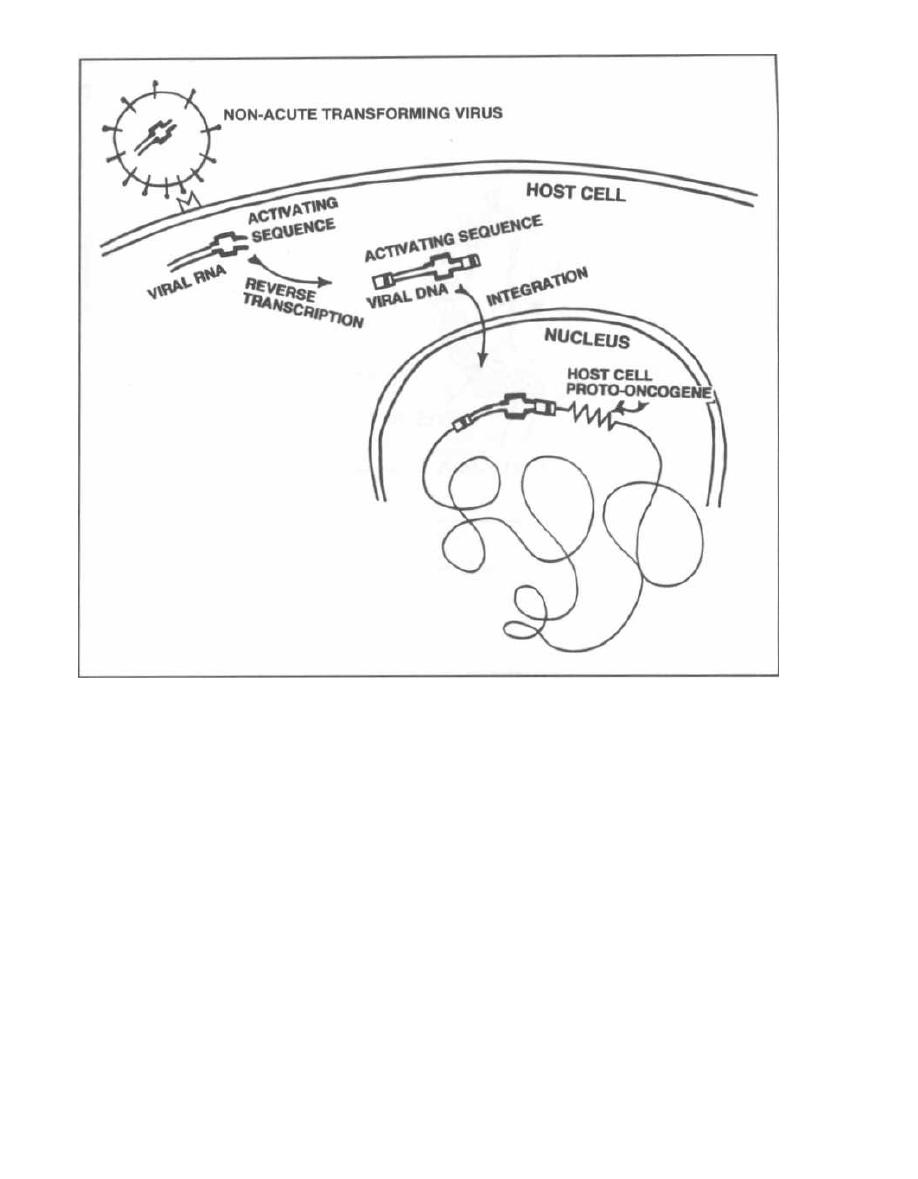
Figure 25-4
To examine this structure fully, we will start from the
i mide and work outward:
1) At the virion core lie 2 identical SS RNA pieces
(a dimer). Associated with these are nucleocapsid
(NC) proteins bound to the RNA and the 3 essen-
tial retroviral enzymes, protease, reverse transcrip-
tase and integrase.
2) Surrounding the RNA dimer lies the capsid shell
which has icosahedral symmetry. The proteins that con-
stitute this shell are called capsid proteins (CA). The
major capsid protein is p24; this can be measured in the
serum to detect early HIV infection.
3) The rest of the virus has the same structure
described for the influenza virus (see Chapter 23). Pro-
teins under the envelope are called matrix proteins.
These proteins serve to hold the glycoprotein spikes
that traverse the lipid bilayer membrane (envelope).
The surface glycoproteins are referred to as gp fol-
lowed by a number: gp 120, and gp 41.
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
193
HN Genome
Fig. 25-6.
The HIV genome (simplified). All retro-
viruses possess, in their RNA genome, two ending long
terminal repeat (LTR) sequences, as well as the
gag
gene,
pol
gene, and
env
gene.
1) LTRs (long terminal repeat sequences) flank
the whole viral genome and serve 2 important functions.
a) Sticky ends: These are the sequences, recog-
nized by integrase, that are involved in insertion
into the host DNA. Transposons, mobile genetic ele-
ments, have similar flanking DNA pieces.
b) Promotor/enhancer function: Once incorpo-
rated into the host DNA, proteins bind to the LTRs
that can modify viral DNA transcription.
2) gag
(Group antigen) sequences code for the pro-
teins inside the envelope: Nucleocapsid (NC), capsid (CA)-
called p24, and matrix (MA) proteins. Thus,
gag
codes for
the virion's major structural proteins that are antigenic.
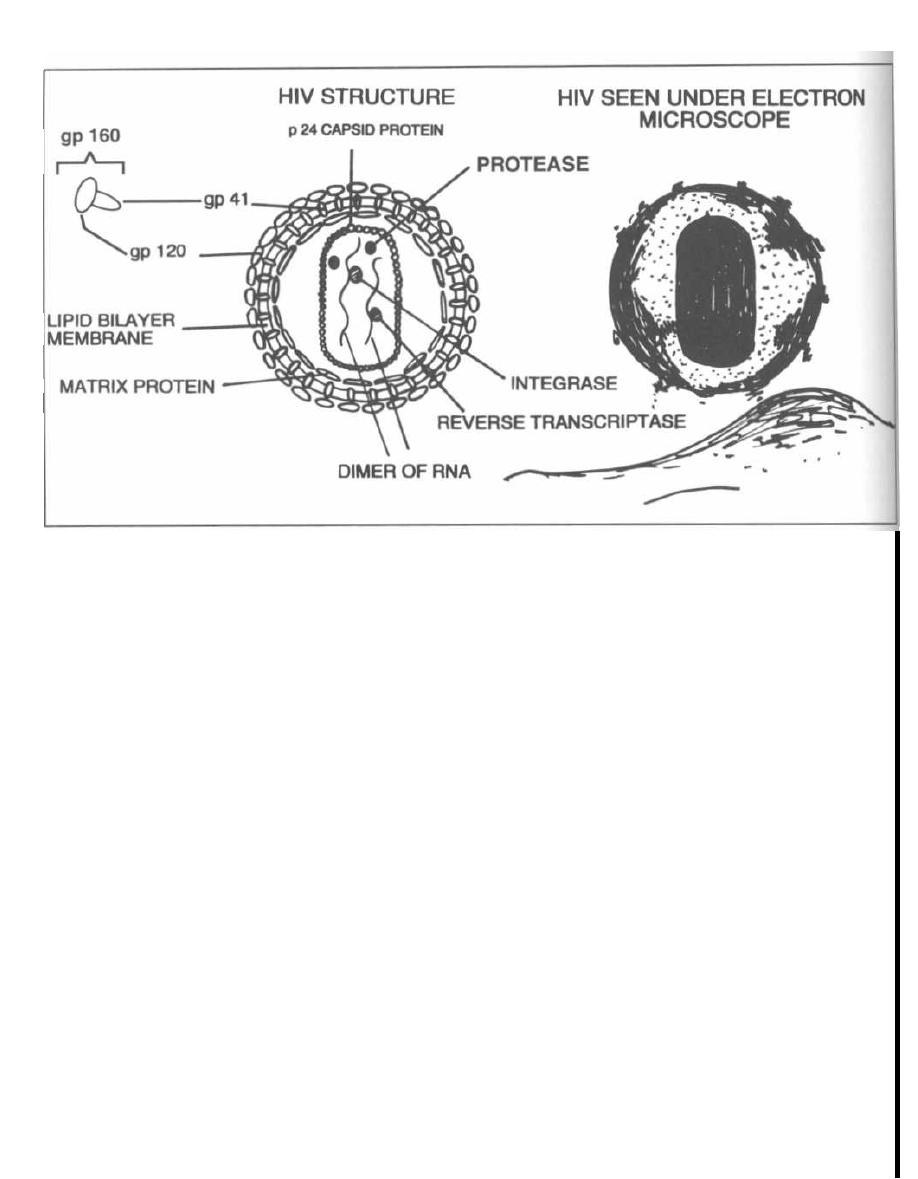
Figure 25-5
( Figure adapted from Fauci-In: Harrison's, 1994; and Haseltine, 1991)
3) pol
encodes the vital protease, integrase, and re-
verse transcriptase enzymes. The only way the retro-
viridae maintain their current POL position in the race
to cause human disease is with these unique enzymes.
Protease is a vital HIV enzyme that cleaves gag and
pol
proteins from their larger precursor molecules (post-
translational modification).
Protease deficient HIV virions can not form their vi-
ral core and are non-infectious. New drugs have been
developed that block the action of the HIV enzyme, pro-
tease. Therapy with these protease inhibitors reduces
HIV levels and increases CD4 T-lymphocyte cell counts.
Similar benefits occur with drugs that inhibit the re-
verse transcriptase enzyme.
4)
env
codes for the ENVelope proteins that, once
glycosylated, form the glycoprotein spikes gp 120 and
gp 41. Gp 120 forms the head and gp 41 the stalk. To-
gether they are called gp 160 and bind to CD4 recep-
tors on T- cells.
Regulatory proteins are encoded by the regulatory
genes:
tat, rev,
and
nef.
Three others
(uif,
vpr,
and
vpu)
have poorly understood actions in vivo and will not
be discussed.
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
194
1) tat
encodes the viral TrAnsacTivator protein.
This protein binds to the viral genome and activates
transcription (thus transactivates). This is a potent pro-
moter of viral activity.
2) rev
is
another promoter that REVS up viral activ-
ity. It achieves this by a unique mechanism:
The HIV virus has multiple reading frames, produc-
ing different mRNAs depending on where splicing oc-
curs. It can be spliced into many pieces, producing the
regulatory proteins such as
tat,
rev, nef
(and others:
uif,
vpr,
and
upu).
Alternatively, it can be spliced only a few
times to produce the major
gag, pol,
and
enu
products
that form the virion.
The
rev
protein binds to the
enu
gene to decrease
splicing. So it REVS up the reading
of gag, pol,
and enu
to produce virions!
3)
nef's
function is uncertain, as experiments have
demonstrated that it can both positively and negatively
regulate HIV expression.
Recent studies have shown that persons infected
with nef deficient HIV-1 do not develop AIDS and do
not suffer T cell destruction. So
nef
may play a critical
role in HIV pathogenesis, and vaccines that target
nef
may be useful.
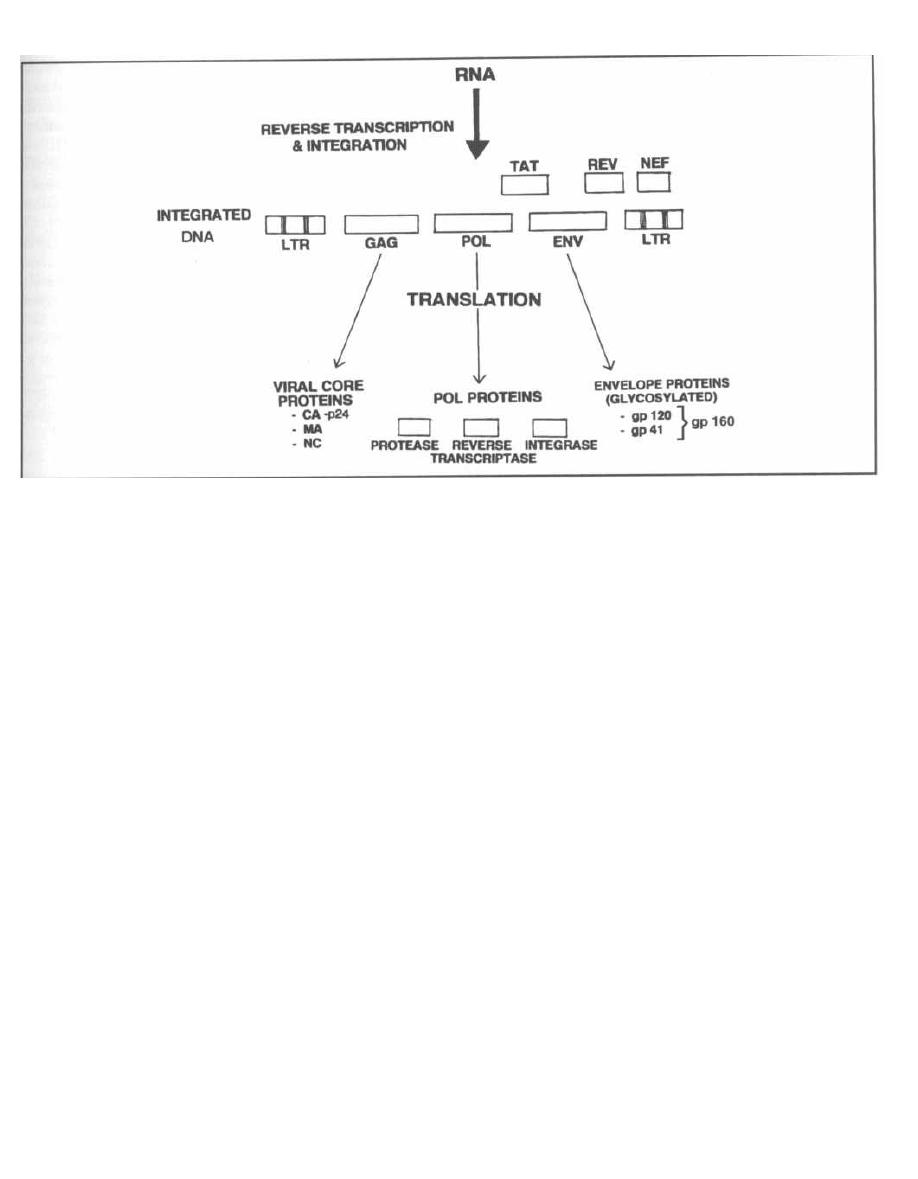
Figure 25-6
Genome Heterogeneity
One of the reasons we are having so much trouble
developing a vaccine against HIV is that it possesses
the ability to change its genome in a critical area.
Within the enu gene, particularly the area encoding the
gp 120 glycoprotein, lie hypervariable regions, where
point mutations occur. In fact, duplications and dele-
tions also occur here, and they occur in multiples of 3
to preserve the codon reading frame! Even the gene
coding for reverse transcriptase undergoes frequent
mutations. In fact, it has one of the highest error rates
(mutation rates) described, leading to HIV strains re-
sistant to zidovudine and other reverse transcriptase
i nhibitor medications. This heterogeneity protects the
virus from the human immune system and vaccine in-
duced antibodies.
HIV INFECTION
Epidemiology and Transmission
As everyone now knows, we are in the midst of a global
pandemic of HIV infection. It is estimated that 47 million
persons worldwide have been infected with the HIV virus
and close to 14 million have died. Ninety percent of in-
fected persons today live in the developing world and most
are in Africa. In fact, it is estimated that in some countries
in Sub-Saharan Africa, one-fourth to one-third of all
adults are infected! The HIV epidemic is rapidly spread-
ing in South and Southeast Asia. While the total number
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
of AIDS cases is on the decline in Western Europe, Aus-
tralia, and the United States, the CDC estimates that
650-900 thousand persons are currently infected with
HIV in the United States.
The disease is transmitted in 2 patterns: 1) In the
Americas and Europe 90% of cases are among homo-
sexuals and IV drug users, resulting in more infected
men then women. 2) In developing areas, namely sub-
Saharan Africa, spread is heterosexual with equal male
and female infection.
The HIV virus is spread by the parenteral route,
much like hepatitis B virus. This occurs with:
1) Sexual activity: Heterosexual and homosexual ac-
tivity is the most common mechanism of transmis-
sion of HIV. HIV is present in seminal fluid as well
as vaginal and cervical secretions. During or following
intercourse, the viral particles penetrate tiny ulcera-
tions in the vaginal, rectal, penile, or urethral mucosa.
Women are 20x more likely than men to get HIV with
vaginal intercourse, likely because of the prolonged ex-
posure of the vagina, cervix, and uterus, to seminal
fluid. Receptive anal intercourse appears to increase the
risk of transmission, likely secondary to mucosal trauma
of the thin rectal wall. Sexually transmitted diseases
also increase the risk of transmission. Organisms such
as Treponema pallidum, herpes simplex virus, Chlamy-
dia trachomatis, and Neisseria gonorrhoeae cause mu-
cosal erosions and may even increase the concentration
of HIV in semen and vaginal fluids. (Inflammation of the
epididymis, urethra, and vaginal mucosa results in an

increase in HIV laden macro-phages and lymphocytes.)
Oral sex is much less likely to result in transmission.
2) Blood product transfusion: HIV can be transmitted
i n whole blood, concentrated red blood cells, platelets,
white blood cells, concentrated clotting factors, and
plasma. Gamma-globulin has not been associated with
transmission. To reduce the risk of transmission via blood
products, blood donors are screened for self reported risk
factors and serologic markers of HIV infection. The latter
i ncludes screening for antibodies to HIV-1 and HIV-2 (by
ELISA) and for p24 antigen. This approach has reduced
the risk of blood product transmission to 1 in 500,000
units transfused.
3) Intravenous drug use with needle sharing: This
has led to growing numbers of infected persons in U.S.
urban centers.
4) Transplacental viral spread from mother to fe-
tus: The rate of transmission is about 30%, and in-
fection occurs transplacentally, during delivery, and
perinatally.
5) Note for students and health care providers: The
risk of contracting HIV from a stick with a needle, con-
taminated with HIV infected blood, is 3 out of a thousand
( 0.3%). The risk is much lower for accidental body fluid
contact with broken skin. There is virtually no risk in
touching an HIV infected patient, unless there is contact
with blood or body fluid. The risk goes up if the injury is
deep, the needle was in a patient's artery or vein, or had
blood visible on it, or if the patient has a high viral load
( MMWR, 1995). To put the risk of transmission of HIV by
needle stick (0.3% transmission risk) into perspective,
the risk of transmission of Hepatitis B virus after a nee-
dle stick from a patient who is Hepatitis B e antigen pos-
itive is about 30%, and for Hepatitis C virus is about 3%.
6) Epidemiologic evidence indicates that the virus
is NOT spread by mosquito bites or casual contact
( kissing, sharing food). There is NO evidence that
saliva, urine, tears, or sweat, can transmit the virus.
Cell Infection
Once the HIV virion is in the bloodstream, its gp 160
(composed at gp 120 and gp 41) glycoproteins bind to the
CD4 receptor on target cells. This CD4 receptor is pres-
ent in high concentration on T-helper lymphocytes.
These cells are actually referred to as CD4+ T-helper
cells. Other cells that possess CD4 receptors in lower
concentrations and which can become infected are
macrophages, monocytes, and central nervous system
dendritic cells. Following HIV binding to the CD4 re-
ceptor, the viral envelope fuses with the infected host
cell, allowing capsid entry.
Part of the mystery of how HIV binds to the CD4 re-
ceptor is as follows. There are two cell surface proteins,
fusin and CKR5, that are produced by T-lymphocytes
and macrophages, respectively. They serve as co-factors
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
19 6
with the CD4 molecule for binding of HIV to lympho-
cytes and macrophages.
Patients who fail to produce normal levels of CKR5
proteins appear to be resistant to HIV infection, and cer-
tain lymphocyte derived proteins (RANTES, MIP1-alpha ,
and MIP1-Beta) that bind to CKR5 apper to inhibit HIV
infection. This now opens the door to new classes of
drugs that block fusin and CKR5! (Feng, 1996; Cohen,
1996; Deng, 1996; Dragic, 1996; Alkhatib, 1996).
The viral RNA is reverse transcribed into DNA in the
cytoplasm. Double-stranded DNA is formed and trans-
ported into the nucleus, where integration into the host
DNA occurs. The integrated DNA may lie latent or may
activate to orchestrate viral replication. There is some
evidence that certain infections, such as with tubercu-
losis, PCP, cytomegalovirus, herpes, Mycoplasma or im-
munizations, will activate T-cells and may promote
viral replication within the T-cells. Stimulation of T-
cells results in production of proteins that bind to the
HIV LTR, promoting viral transcription.
Following viral replication the new capsids form
around the new RNA dimers. The virion buds through
the host cell membrane, stealing portions of the mem-
brane to use as an envelope, leaving the T-cell dead.
Immunology and Pathogenesis
Following initial infection, HIV can begin replication
i mmediately, resulting in rapid progression to AIDS, or
there can be a chronic latent course. The former, most
common pattern occurs in 3 stages starting with initial
i nfection, marked by an acute mononucleosis-like viral
illness. This progresses for a variable number of years
( median 8 but range of less than 1 to greater than 20) of
disease-free latency. After AIDS develops most patients
die within 2 years if they do not receive effective anti-
retroviral therapy ( Fig. 25-7).
1) An acute viral illness like mononucleosis (fever,
malaise, lymphadenopathy, pharyngitis, etc.) develops
i n 80% about 1 month after initial exposure. There are
high levels of blood-borne HIV ( viremia) at this stage,
and the viruses spread to infect lymph nodes and
macrophages. An HIV-specific immune response arises,
resulting in decreased viremia and resolution of the
above symptoms. However, HIV replication continues
i n lymph nodes and peripheral blood.
2) A clinical latency follows for a median of 8 years
during which there are no symptoms of AIDS, although
some patients develop a dramatic generalized lym-
phadenopathy (possibly secondary to an aggressive im-
mune attack against HIV harbored in the lymph nodes).
This is not a true viral latency without viral replication;
HIV continues to replicate in the lymphoid tissue and
there is a steady gradual destruction of CD4 T-lym-
phocytes (helper) cells. CD4+ T-helper cells are the

number one target of HIV. The virus reproduces in
these cells and destroys them.
Toward the end of the 8 years, patients are more sus-
ceptible to bacterial and skin infections, and can de-
velop constitutional (systemic) symptoms such as fever,
weight loss, night sweats, and adenopathy.
3) AIDS develops for a median of 2 years followed by
death. AIDS is now defined as having a CD4 T-lympho-
cyte count of less than 200 (with serologic evidence
of HIV infection such as a positive ELISA or west-
ern blot test) and/or one of many AIDS-defining oppor-
tunistic infections, which are infections that usually only
patients with AIDS develop. These include Candida
esophagitis, Pneumocystis carinii pneumonia, the malig-
nancy Kaposi's sarcoma, and many others.
Fig. 25.7. The clinical course of HIV infection
(acute viral illness, clinical latency, and AIDS) as CD4+
T-cells decline over time, and the opportunistic infec-
tions that develop at specific CD4 T-cell counts.
1) Normal CD4+ T-cell counts are 1000 cells/µl blood.
In HIV-infected persons the count declines by about 60
cells/ml blood/year.
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
2) CD4+ T-cell count of 400-200 (about 7 years):
Constitutional symptoms (weight loss, fever, night
sweats, adenopathy) develop as well as annoying skin
infections, such as severe athlete's foot, oral thrush
(Candida albicans), and herpes zoster. Bacterial infec-
tions, especially Mycobacterium tuberculosis, become
more common as CD4 counts drop below 400!!
3) CD4+ T-cell count less than 200 (about 8 years):
As the immune system fails, the serious opportunistic
killers set in, such as Pneumocystis carinii pneumonia,
Cryptococcus neoformans, and Toxoplasma gondii.
4) CD4+ T-cell count less than 50: At this level the
immune system is almost completely down. Mycobac-
terium avium-intracellulare, normally only causing in-
fection in birds, causes disseminated disease in the
AIDS patients. Cytomegalovirus infections also rise as
the count moves from 50 to zero.
Viral Load
CD4 counts are used to determine severity of HIV in-
fection, risk of opportunistic infection, prognosis, and re-
sponse to anti-viral therapy. We can now measure plasma
HIV RNA by the polymerase chain reaction (PCR) or

branched chain DNA assay. There is mounting evidence
that higher plasma HIV RNA levels ( viral load) correlate
with a greater risk of opportunistic infection, progression
to AIDS, and risk of death (Mellors,1996; Galetto-Lacour,
1996). CD4 counts are still the best predictor of a patient's
current risk for particular opportunistic infections. David
Ho has popularized the train analogy to explain the pre-
dictive values of HIV viral load and CD4 counts. Viral load
tells you the speed at which the train is heading for the
cliff (low CD4, development of opportunistic infections
and death) while the CD4 count tells you where the train
currently is! For example, if a patient has a CD4 count of
450 cells/mL and a viral load of >10
6
copies/µL that pa-
tient is at low risk for developing Pneumocystis carinii
pneumonia today (CD4>200 cells/micL ) but is at great risk
in the future for rapid CD4 count decline, opportunistic in-
fection and death if not treated.
Mechanism of T-Cell Death
The CD4 receptor appears to be involved in T-cell
death. Monocytes and macrophages, which possess
lower CD4 receptor concentrations, are not destroyed as
extensively as are T-cells.
When a T-helper cell is infected, and the virus produces
its structural proteins, gp 160 (composed of gp 120 and gp
41) is integrated into the T-helper cell cytoplasmic mem-
brane. The virion will bud at the site of gp 160 insertion,
stealing this portion of the membrane to form its envelope.
Three mechanisms ofT-cell death have been observed:
1) When the virion is budding, the gp 160 (in the T-
cell membrane) may bind to adjacent CD4 receptors on
the same T-helper cell membrane, tearing the T-cell
membrane and destroying the cell.
2) A second phenomenon occurs between infected
cells and noninfected CD4 cells. The gp 160 in the in-
fected cells binds to other CD4 T-helper cells, resulting
in cell-to-cell fusion. One infected cell can fuse with as
many as 500 uninfected CD4+ T-helper cells, forming
multinucleated giant cells.
3) Gp 160 in the T-cell membrane may mark the cell
as non-self, resulting in autoimmune T-cell destruction
by cytotoxic CD8 T lymphocytes.
HIV probably also kills T-cells directly by inhibiting
host cell protein synthesis.
What Is the Clinical Significance
of These Events?
T-Cell death: A healthy person has about 1000
CD4+ T-helper cells per milliliter of blood. HIV-induced
T-cell death results in a decline of about 60 CD4+ T-
cells/µl a year. The T-helper cell plays a vital role in the
orchestration of the cell-mediated immune system, acti-
vating and recruiting macrophages, neutrophils, B-lym-
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
198
phocytes, and other immune system effectors. A severe
i mmunodeficiency state follows the loss of CD4+ T-cells.
Multinucleated giant cells: This T-cell to T-cell fu-
sion allows the virus to pass from an infected cell to an
uninfected cell without contacting the blood. This
may protect the virus from circulating antibodies.
B-Lymphocytes: HIV does not actually infect the B-
cells; however, B-cell dysfunction does occur with HIV
i nfection. There is a polyclonal activation of B-cells, re-
sulting in an outpouring of immunoglobulins. This
hypergammaglobulinemia results in immune-com-
plex formation and autoantibody production. The most
i mportant dysfunction that occurs is a diminished abil-
ity to produce antibodies in response to new antigens or
i mmunization. This is very serious in infants with AIDS
because they cannot develop humoral immunity to the
vast number of new antigens they are exposed to.
Monocytes and macrophages: HIV infects these
cells and actively divides within them. However, these
cells are not destroyed by HIV. This is clinically signifi-
cant in 2 ways:
1) Monocytes and macrophages serve as reservoirs
for HIV as it replicates, protected within these cells
from the immune system.
2) These cells migrate across the blood-brain barrier,
carrying HIV to the central nervous system. HIV causes
brain disease, and the predominant cell type harboring
HIV in the central nervous system is the monocyte-
macrophage line.
BIG PICTURE: HN infection diminishes CD4
T-lymphocyte (helper) cell numbers and function.
The CD4 T-helper cells are involved in all immune
responses. So all immune cells have some kind of
altered function. As T-cell numbers decline, the
host becomes susceptible to unusual infections
and malignancies that normally are easily con-
trolled by an intact immune system.
ACQUIRED IMMUNODEFICIENCY
SYNDROME (AIDS)
Fig. 25-8.
The acquired immunodeficiency syndrome
(AIDS) is an extremely complex disease. To better un-
derstand this complexity, consider 2 processes that oc-
cur. The HIV virus causes 1) direct viral disease and 2)
disease secondary to the immunodeficiency state.
1) Direct viral disease
Constitutional (widespread body) symptoms
Neurologic damage
2) Disease secondary to the immunodeficiency state
Failure of the immune surveillance system that
prevents malignancies
Secondary infections by pathogens and normal
flora (opportunistic infections)
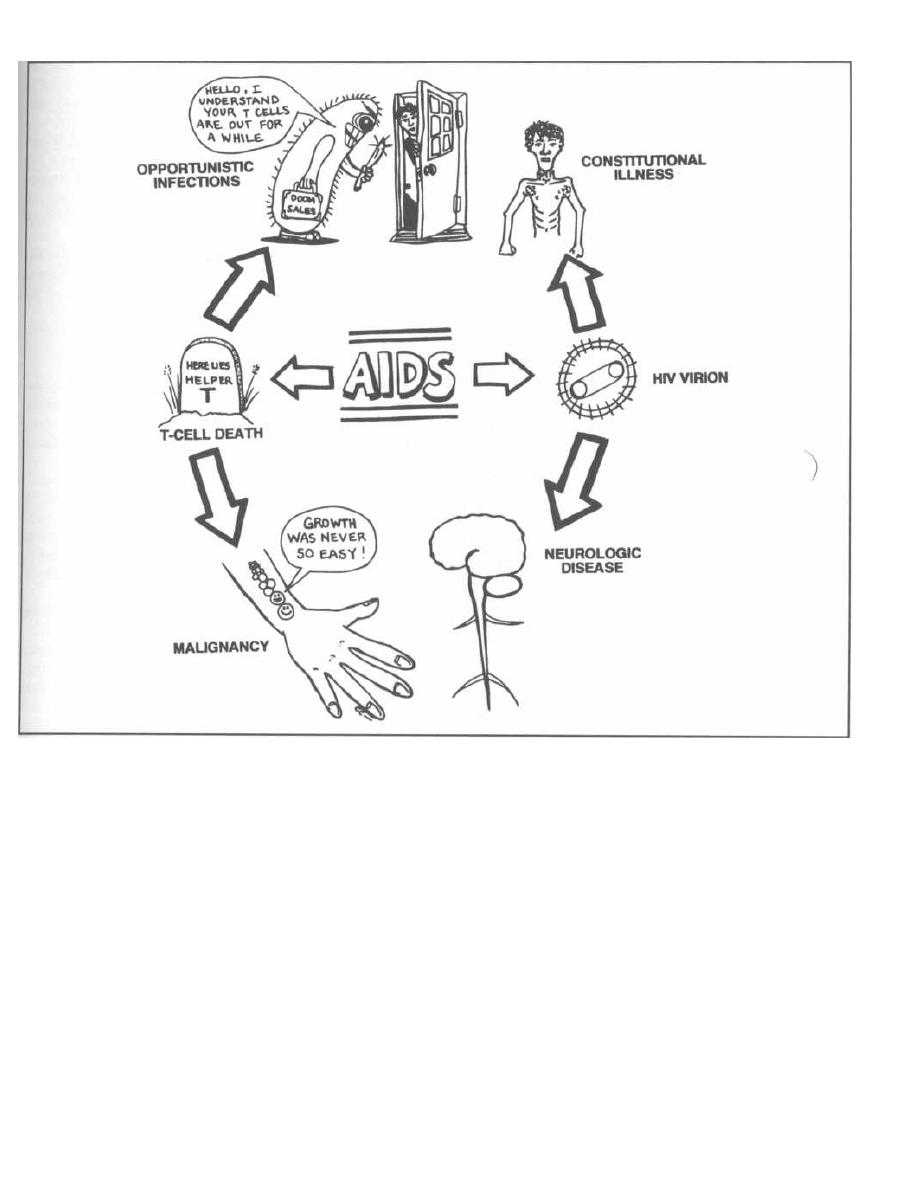
Figure 25-8
Constitutional Illness
AIDS patients suffer from night sweats, fevers, en-
larged lymph nodes, and severe weight loss. The weight
loss is often referred to as the wasting syndrome.
Neurologic Disease
The HIV virus is carried to the central nervous sys-
tem by the monocyte-macrophage cells. It is unclear at
this time whether the neuronal damage is caused by the
inhibition of neuronal growth by the HIV envelope
proteins or an autoimmune damage caused by the in-
fected monocyte-macrophages themselves.
Many patients with HIV infection suffer from some
form of neurological dysfunction. The brain can suffer
diffuse damage (encephalopathy) resulting in a pro-
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
199
gressive decline in cognitive function referred to as the
AIDS dementia complex. Meningeal infection results in
aseptic meningitis. The spinal cord can become infected,
resulting in myelopathy, and peripheral nerve involve-
ment results in a neuropathy.
Malignancies
AIDS patients suffer from a high incidence of B-cell
lymphoma, often presenting as a brain mass. Half of
B-cell lymphomas in AIDS patients are found to contain
Epstein-Barr virus DNA.
Another common AIDS associated malignancy is
Kaposi's sarcoma. Most cases of Kaposi's sarcoma
(96%) occur in homosexual men, which suggests that
there may be a co-factor, which appears to be a new
herpes virus called HHV-8. HHV-8 DNA sequences

have been found in Kaposi's sarcoma, and antibodies
to HHV-8 are found in high concentrations in most pa-
tients (80%) with Kaposi's sarcoma and in 35% of ho-
mosexual HIV positive men (Moore, 1995; Kedes,
1996; Gao, 1996). The lesions are red to purple,
plaques or nodules, and arise on the skin all over the
body. The course can range from nonaggressive dis-
ease, with limited spread and only skin involvement,
to an aggressive process involving skin, lymph nodes,
l ungs, and GI tract.
Opportunistic Infections
The most common manifestation of AIDS is the sec-
ondary infection by opportunists. These are bugs that
are normally pushovers to the intact immune system
but wreak havoc in the absence of T-helper defenses (see
Fig. 25-7.).
Bacterial Infections
AIDS patients often have many permanent in-
dwelling intravenous lines or are in the hospital with
central venous lines. These serve as entry points for bac-
teremia caused by
Staphylococcus aureus
or
Staphylo-
coccus epidermidis.
The poorly functioning B-cells and their impaired hu-
moral immunity result in more infections with encap-
sulated organisms such as
Haemophilus influenzae
and
Streptococcus pneumoniae.
Mycobacterium tuberculosis:
Remember the piv-
otal role of cell-mediated immunity in defense against
Mycobacterium tuberculosis.
AIDS patients have a
higher chance of tuberculosis reactivation (about 10%
chance per year). (More on page 105).
Mycobacterium
avium-intracellulare
(MAI):
This atypical mycobacterium, also called
Mycobacterium
avium-complex (MAC),
can be isolated from many sites
( GI tract, liver, bone marrow, lymph nodes, lungs, blood)
in infected patients. It causes a smoldering, wasting dis-
ease characterized by fever, night sweats, weight loss,
and often diarrhea (GI tract infection) and elevated liver
function tests. (More on page 106).
Fungal Infections
Candida albicans:
This yeast is very common in
HIV-infected patients. It causes oral thrush and
esophagitis. Thrush looks like white plaques on the oral
mucosa and when scraped off with a tongue blade leaves
a red bleeding base. (More on page 150).
Cryptococcus neoformans:
This fungus causes a
meningitis in about 10% of AIDS patients. Fever, nau-
sea, and vomiting may hint at cryptococcal meningitis.
Important: AIDS patients are similar to the elderly and
children: Without a full immune system they often
do not exhibit meningeal inflammation. Only 25%
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
200
of AIDS patients with cryptococcal meningitis will pre-
sent
with
headache,
mental status changes, or
meningeal signs. For example: A normal host with
meningitis would have meningeal inflammation with
meningismus (positive Kernig's and Brudzinski's sign,
stiff neck, headache). An AIDS patient can have a rag-
i ng meningitis with only fever. You must have a high
level of suspicion and always consider doing a lumbar
puncture, for cerebrospinal fluid testing, on AIDS pa-
tients with fever. (More on page 149).
Histoplasma capsulatum
and
Coccidioides im
-
mitis:
These fungi produce disseminated disease in
AIDS patients, infecting meninges, lungs, skin, and
other areas. (More on page 147).
Viral Infections
Herpes zoster: Painful vesicles develop in der-
matomal distribution as the varicella-zoster virus ven-
tures forth from its latency in the dorsal sensory
ganglia. AIDS patients can also develop disseminated
non-dermatomal zoster.
Epstein-Barr virus, another herpes family virus, is
thought to cause oral hairy leukoplakia (OHL). OHL
usually develops when CD4 counts are <400 and is
characterized by white hairlike projections arising from
the side of the tongue. This is differentiated from Can-
didal thrush by the fact that OHL will not rub off
with
a tongue blade.
Herpes simplex: Viral infection results in severe
genital and oral outbreaks.
Cytomegalovirus (CMV): This virus can cause
chorioretinitis and blindness. Visual compromise must
be looked into carefully in AIDS patients because it can
represent the retinal lesions of CMV or brain masses of
toxoplasmosis and lymphoma. CMV can also cause
esophagitis (pain with swallowing) and diarrhea.
Protozoal Infections
Pneumocystis carinii
pneumonia (PCP): This is
the most common opportunistic infection. Without pro-
phylactic treatment there is a 15% chance each year
of infection when the CD4+ T-cell count is below
200. AIDS patients who develop PCP have cough and
hypoxia. The chest X-ray can be normal or show an in-
terstitial infiltrates. Pneumothorax complicates 2% of
PCP cases. About 80% of AIDS patients will get this at
least once in their lifetime unless prophylactic antibi-
otics are taken. (More on page 235).
Toxoplasma gondii:
This parasite causes mass le-
sions in the brain in 15% of AIDS patients. Patients
present with fever, headache, and focal neurologic
deficits (seizure, weakness, aphasia). A CT scan will
show contrast-enhancing masses in the brain. (More on
page 234).

Cryptosporidium, Microsporidia, and Isospora
belli.
These parasites cause chronic diarrhea in
patients with AIDS. (More on page 231).
DIAGNOSIS of HIV and AIDS
Following infection with HIV, viral RNA or antigens
(such as p24) can be detected in the blood within weeks.
Three to 6 weeks later antibodies against HIV antigens
appear. The enzyme-linked immunosorbent assay
(ELISA) test detects antibodies. This test is very sensi-
tive at detecting HIV infection (sensitivity of 99.5%) but
it often gives false positive results.
To decrease this rate of false positives, a second
ELISA is recommended on the original sample and if
this is positive again, do a western blot test. In this
test, HIV antigens ( gag, pol, and enu proteins) are
separated in bands on paper by molecular weight. The
person's serum is then added to this paper. If the
serum contains antibodies against HIV antigens they
will stick to the antigens on the paper. Lastly, anti-
human antibodies (labeled with enzymes) are added;
these stick to the antibodies on the antigens, lighting
up "bands" on the paper. A western blot is considered
positive if it has bands to 2 HIV gene products (p24,
gp41, gp120) (see Figs. 25-5 and 25-6).
Direct viral culture in cell lines, p24 antigen capture,
and polymerase chain reaction (PCR) and BDNA are
used to identify HIV whole virus, p24 antigen, and
DNA/RNA respectively.
HN infection should be suspected when an at-risk
individual (homosexual, IV drug user, sexual partner
of an at-risk individual, etc.) develops constitutional
symptoms such as fevers, night sweats, and generalized
adenopathy, or suffers from recurrent bacterial infec-
tions, tuberculosis, skin zoster or tinea infections, or
oral thrush ( Candida).
AIDS is diagnosed when the CD4 T-lymphocyte count
is less than 200 (with serologic evidence of HIV infection
such as a positive ELISA or western blot test) and/or the
patient has one of many AIDS-defining opportunistic in-
fections, which are infections that usually only patients
with AIDS develop. These include Candida esophagitis,
Pneumocystis carinii pneumonia, the malignancy Ka-
posi's sarcoma, and many others.
CONTROL, TREATMENT, CURE?
Efforts directed toward viral control are moving along
4 lines:
1) Prevention of HIV viral infection.
2) Vaccine development.
3) Limiting growth of HIV, once infection has occurred.
4) Treating the opportunistic infections that ulti-
mately cause death.
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
201
Prevention
Education to avoid high-risk activities (needle
sharing, multiple sexual partners, unprotected sex).
Screening blood products with ELISA and p24
antigen.
Vaccine Development
The goal of vaccination is to stimulate an immune re-
sponse that will counter a subsequent infection. Most
vaccines stimulate an antibody response to a viral anti-
gen, resulting in the neutralization of the virus. Persons
infected with HIV develop antibodies against HIV de-
terminants. Early in HIV infection antibodies arise that
bind to a hypervariable portion of the envelope glyco-
protein gp 120, called the
V3
loop. These V3 specific
antibodies will neutralize only the exact strain of HIV
that elicited the antibody. Chimpanzees given V3 neu-
tralizing monoclonal antibody (passive immunization)
and chimpanzees actively immunized with the V3 enve-
lope glycoprotein were protected from injected cell-free
HIV virus of the same strain. These vaccines only pro-
tect against a virus with the exact same V3 hypervari-
able region and only during peak immunity.
Another later developing antibody response occurs
against the CD4 binding domain (gp 160, composed
of gp 120 and gp 41); this domain is responsible for
binding to T-lymphocyte CD4 receptors. The CD4 bind-
ing domain antigen is more conserved, meaning that it
is similar in many HIV strains. Antibodies against this
domain will prevent viral binding to the CD4 receptor,
neutralizing HIV-1 in vitro.
Since the above antibody responses develop with HIV
infection and with all the ongoing efforts to develop a
vaccine, why are we told that successful vaccination is a
distant reality?!?
There are many challenges to the development of a
successful vaccine against HIV-1:
1) Rapid mutation: HIV envelope glycoproteins
mutate rapidly, so there are many different strains. The
rapidly mutating V3 loop of gp 120 and the reverse tran-
scriptase enzyme combined with rapid viral reproduc-
tion over a long disease course results in different
"quasi-species," even in the same person. A vaccine
would need to target a conserved region like the CD4
binding domain.
2) HIV is transmitted from cell-to-cell: With
syncytial giant-cell formation, HIV is able to pass
from one cell to another without contacting the blood-
stream that carries antibodies. The virus can thus
escape the antibody-mediated or humoral immune
system. Protection against HIV infection also requires
cell-mediated immunity: HIV proteins in an infected
cell are expressed on the cell surface associated with
class I molecules of the major histocompatibility

complex. Circulating HIV-specific cytotoxic T-lympho-
cytes will recognize this complex and lyse the cell, de-
stroying HIV inside. This cell-mediated response does
occur in patients with HIV infection, and a successful
vaccine would have to stimulate this arm of the im-
mune system.
3) Poor animal model: One would like an animal
model that is easily obtainable, cheap, and would develop
an AIDS-like illness when infected with HIV. There is no
such model. The only species that can be infected are the
great apes; they are expensive, scarce, and do not get
AIDS when infected with HIV. However, their antibody
response to HIV can be followed, and they do get an
AIDS-like illness when infected with SIV (which shares
sequence homology with HIV-2).
Current Vaccine Research Efforts
1) Live viruses can be altered or attenuated so they
lose their virulence while still stimulating immunity
(e.g., polio and measles vaccine). This type of vaccine
would stimulate cellular and humoral immunity, as
well as mucosal immunity. Studies with live viruses
have used SIV and HIV-2, which have been made non-
pathogenic secondary to gene deletions. The limitation
of this approach involves the danger of new mutations
occurring in the non-pathogenic live virus that might
make it pathogenic. Also, the great apes do not develop
an AIDS-like disease, so how can we be sure such a vac-
cine is non-pathogenic?
2) A recombinant HIV-1 envelope glycoprotein
vaccine can be made by splicing the HIV gene, which
codes for envelope glycoprotein antigens
( V3
loop, CD4
binding domain, or others), into the DNA of tumor-cell
lines. The tumor cells will produce the HIV envelope
glycoprotein in mass quantities that can be used as a
vaccine. The hepatitis B vaccine is made in this manner,
with cell lines producing the hepatitis B surface antigen
( HBsAg). As mentioned above, however, these vaccines
only protect against a virus with the exact same anti-
genic region used in the vaccine and only during peak
i mmunity. They also fail to activate a cytotoxic T-lym-
phocyte response.
3) Live recombinant organisms can be used to
carry and express HIV genes. The vaccinia virus (live
attenuated virus used for smallpox vaccine), bacille Cal-
mette-Guerin (bacteria used for
Mycobacterium tuber-
culosis
vaccine), and adenovirus strains are some of the
organisms used as vectors in vaccine experiments. Some
studies have demonstrated an antibody and T-lympho-
cyte response to proteins produced by these recombi-
nant organisms.
4) Direct intramuscular injection of HIV genes
may elicit humoral and cell-mediated immune responses.
5) Soluble CD4 receptors delivered to cultured cells
block HIV infection. The CD4 receptors bind to HIV gp 120,
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
202
thus preventing HIV from binding to the CD4+ T-helper
cells. Simian immunodeficiency virus (SIV), a close rela
-
tive to HIV-2, infects primates. Monkeys with SN were
given soluble CD4 with improvement in their disease (in-
creased T-lymphocyte counts and reduced viral load),
( Letvin, 1993)
For further reading refer to: Graham BS, Wright PF.
Drug therapy: Candidate AIDS vaccines. N Engl J Med
1995;333:1331-9.
Limiting Viral Growth
Triple drug therapies-Highly Active Antiretroviral
Therapy (HAART) have been used to bolster the im-
mune system in HIV-positive and AIDS patients,
which has decreased the rate of development of oppor-
tunistic infections, including
Mycobacterium avium,
CMV retinitis, & oropharyngeal candidiasis. Please see
Chapter 29, the anti-viral antibiotic chapter, which ex
-
tensively discusses current anti-HIV drug strategies.
Treating the Opportunistic infections
1) Pneumocystis carinii
pneumonia
(PCP):
Trimethoprim and sulfamethoxazole are given prophy-
lactically when CD4+ T-cell counts drop below
200-250. Greater than 90% of PCP infections are being
prevented with this prophylactic intervention!
2) Toxoplasmosis: Brain lesions are treated with
another tetrahydrofolate reductase inhibitor/sulfa com-
bination called pyrimethamine/sulfadiazine. Patients
i mprove rapidly. In fact, if there is no brain mass
shrinkage (as seen by CT scan) by 2-3 weeks, then the
diagnosis of toxoplasmosis is unlikely. Brain biopsy
should then be done to determine whether the mass is a
B-cell lymphoma.
The same medicine (trimethoprim and sulfamethox-
azole), used for PCP prophylaxis, also prevents toxo-
plasmosis! It prevents two birds with one stone.
3) Mycobacterium tuberculosis
and
Mycobac-
terium avium-intracellulare:
Treatment of tubercu-
losis is covered in Chapter 15.
Azithromycin or clarithromycin can be given
daily for prophylaxis against future MAI infections.
4) CMV: Treatment with ganciclovir or foscarnet can
prevent progression of visual loss.
5) Herpes, Varicella-zoster: Acyclovir.
6) Candida albicans:
Oral clotrimazole, nystatin,
or fluconazole preparations for thrush and esophagitis.
Systemic fungal infections are treated with intravenous
amphotericin B or fluconazole.
7) Bacteria: Appropriate antibiotics.
AIDS patients are now surviving for prolonged peri-
ods with CD4+ T-cell counts approaching zero. They are
often on more than 10 different medications.

A Final Word
AIDS is a disease that has no dignity, a disease that
cripples the immune system, allowing the scourge
of
all
infestations. You are becoming a physician in the dawn
of
a new epidemic, and you will certainly play a role in
the control
of
this epidemic.
References
Alkhatib G, et al. CC-CKR5: a RANTES, MIP-la, MIP-1 beta
receptor as a fusin cofactor for macrophage-trophic HIV-1.
Science 1996;272:1955-8.
Case-control study of HIV seroconversion in health-care work-
ers after percutaneous exposure to H1V-infected blood-
France, United Kingdom, and United States, January
1988-August 1994. MMWR 1995;44:929-33.
Cohen J. HIV cofactor found. Science 1996;272:809-10.
Deng H, et al. Identification of a major co-receptor for primary
isolates of HIV-1. Nature 1996;381:661-6.
Dragic T, et al. HIV-1 entry into CD4 T cells is mediated by the
chemokine receptor CC-CKR5. Nature 1996;381:667-73.
Fauci AS, Lane HC. Human immunodeficiency virus (HIV)
disease: AIDS and related disorders. In: Isselbacher, Braun-
wald, et al., eds. Harrison's Principles of Internal Medicine.
13th ed. New York: McGraw-Hill, 1994;1566-1618.
Feng Y, et al. HIV-1 entry cofactor: functional CDNA cloning
of a seven-transmembrane, G protein-coupled receptor. Sci-
ence 1996;272:872-7.
Galetto-Lacour A, et al. Prognostic value of viremia in patients
with long-standing human immunodeficiency virus infec-
tion. J Infect Dis 1996;173:1388-93.
Gao SJ, et al. KSHV antibodies among Americans, Italians,
and Ugandans with and without Kaposi's sarcoma. Nature
Medicine 1996;2:925-8.
Graham BS, Wright PF. Drug therapy: candidate AIDS vac-
cines. N Engl J Med 1995;333:1331-9.
CHAPTER 25. RETROVIRIDAE, HIV, AND AIDS
203
Haseltine WA. Molecular biology of the human immunodefi-
ciency virus type 1. The FASEB journal 1991:2349-2360.
Kedes DH, et al. The seroepidemiology of human herpes virus
8 (Kaposi's sarcoma-associated herpesvirus): distribution of
infection in KS risk groups and evidence for sexual trans-
mission. Nature Medicine 1996;2:925-8.
Lackritz EM, Satten GA, et al. Estimated risk of transmission
of the human immunodeficiency virus by screened blood in
the United States. N Engl J Med 1995;333:1721-5.
Letvin NL. Vaccines Against Human Immunodeficiency Virus-
Progress and Prospects. N Engl J Med 1993;329:1400-1405.
Mellors JW, et al. Prognosis in HIV-1 infection predicted by
the quantity of virus in plasma. Science 1996;272:1167-70.
Moore PS, Chang Y. Detection of herpesvirus-like DNA se-
quences in Kaposi's sarcoma in patients with and those
without HIV infection. N Engl J Med 1995;332:1181-5.
Pantaleo G, Graziosi C, Fauci A. The immunopathogenesis of
human immunodeficency virus infection. N Engl J Med
1993;328:327-335.
Watkins BA, Klotman MF, Gallo RC. Human immunodefi-
ciency viruses. In: Mandell
Bennett VF, Polin R, eds.
Principles and Practice of Infectious Diseases. 4th edition.
New York: Churchill Livingstone 1995;1590-1606.
Recommended Review Articles:
Esparza J and Bhamarapravati N. Accelerating the develop-
ment and future availability of HIV-1 vaccines: why, when,
where, and how? Lancet 2000;355:2061-2066.
Harrington M and Carpenter CC. Hit HIV-1 hard, but only
when necessary. Lancet 2000;355:2147-2152.
Kovacs JA and Masur H. Prophylaxis against opportunistic in-
fections in patients with human immunodeficiency virus in-
fection. N Engl J Med 2000;342:1416-1429.

CHAPTER 26. HERPESVIRIDAE
Figure 26-1
We all probably have at least 1 of the herpes family
viruses living in a latent state in our bodies right now!
Even before entering medicine we have seen people with
herpesviridae infections. People with "fever blisters" of
HSV-1, the child with multiple blisters covering the
body with chickenpox (varicella-zoster), and the teenage
friend who had to miss school with mononucleosis
(Epstein-Barr virus).
There are some generalities that the herpesviridae
share:
1) They can develop a latent state.
2) The members in the sub-family alpha have a cyto-
pathic effect on cells, which become multinucleated
giant syncytial cells with intranuclear inclusion
bodies.
3) Herpesviridae are held at bay by the cell-medi-
ated immune response.
Latency: During the primary infection the viruses
migrate up the nerves to the sensory ganglia and reside
there. The viruses rest there until reactivation occurs
through some stress, such as menstruation, anxiety
states, fever, sunlight exposure, and weakening of the
cell-mediated immune system, as with AIDS or chronic
disc ase. The viruses then migrate out to the peripheral
skin via the nerves to cause local destruction.
Fig. 26-1.
Captain Herpes hiding in latency in his
dorsal sensory ganglia fortress.
Cytopathic effect: The herpesviridae that cause cell
destruction are the alpha sub group viruses (herpes
simplex virus 1 and 2, and varicella-zoster). This cell de-
struction results in the separation of the epithelium and
causes blisters (vesicles). Microscopic study of skin
biopsies or scrapings from blister bases in herpes sim-
plex, chickenpox, and zoster all reveal multinucleated
giant cells and intranuclear inclusion bodies. Viral pro-
teins are inserted into the host cell plasma membranes,
resulting in cell fusion to form multinucleated giant
cells. Intranuclear inclusions are considered to be areas
of viral assembly.
204
Both CMV (beta subgroup) and Epstein-Barr virus
(gamma subgroup) have less cytopathic effects.
Patients with compromised cell-mediated im-
mune status are more likely to suffer from severe her-
pesviridae infections such as disseminated HSV or
multi-dermatomal zoster. CMV frequently causes dis-
ease in AIDS patients.
Herpes Simplex Virus 1 (HSV-1)
Antibodies to HSV-1 are present in 90% of adults by
the fourth decade, with those from lower socioeconomic
classes being more likely to acquire HSV earlier. Most
primary infections are not even noticed. In fact, fewer
than 1% are clinically apparent. When there are clinical
symptoms, patients will present with:
1) Gingivostomatitis: Painful swollen gums and
mucous membranes with multiple vesicles. Fever and
systemic symptoms can accompany the infection, and
the disease will resolve in about 2 weeks. Vesicles can
also appear on areas of the skin where viral entry has
occurred.
2) Reactivation: About one fourth of previously in-
fected
people
have reactivation infection during
stressed states. AIDS patients can present with severe
reactivation of HSV.
3) Herpetic keratitis: the most common infectious
cause of corneal blindness in the United States.
4) Encephalitis: HSV-1 is the most common cause
of viral encephalitis in the U.S. Infection of the brain
cells occurs, with cell death and brain tissue swelling.
Patients present with sudden onset of fever and focal
neurological abnormalities. HSV-1 must always be
considered, because herpes is one of the few treatable
causes of viral encephalitis!!
Herpes Simplex Virus 2 (HSV-2)
HSV-2 is antigenically distinct from HSV-1. It com-
monly causes genital disease that is sexually trans-
mitted. However, HSV-2 can also cause oral/skin/eye

disease, and HSV-1 can cause genital herpes. The dif-
ference is unimportant clinically because both can cause
the same symptoms, and the treatment is the same. Pa-
tients with genital herpes get vesicles on the vagina,
cervix, vulva, perineum, and glans and shaft of the pe-
nis. The vesicles are painful, with burning and itching,
often associated with urination.
Neonatal Herpes
HSV infection during pregnancy can result in
transplacental viral transfer. The infection of the fetus
can cause congenital defects or intrauterine death. The
neonate can also acquire the illness during delivery if
the mother is having an active genital infection.
Figure 26-2
Fig. 26-2.
Death, carrying his TORCH, visits the
pregnant mother and her baby. Remember that herpes
is one of the TORCHES organisms that can cross the
blood-placenta barrier:
T0: Toxoplasmosis
R:
Rubella
C:
Cytomegalovirus
HE: HErpes, HIV
S:
Syphilis
Varicella-Zoster Virus (VZV)
As the name implies, this virus causes 2 diseases:
varicella (chickenpox ) and herpes zoster (shingles).
Chickenpox is not caused by the pox viridae!!! Varicella
is usually a disease of children. After resolution the
virus remains latent as described previously. Later in
life, reactivation can cause the second disease, zoster.
Once again, with stressors or depressed cell-mediated
immunity (usually in the elderly), the virus will migrate
CHAPTER 26. HERPESVIRIDAE
20 5
out along sensory nerve paths and cause vesicles simi-
lar to those of chickenpox. However, with this reactiva-
tion infection, the vesicles appear in a dermatomal
distribution, almost always unilaterally.
Varicella
( Chickenpox)
VZV is highly contagious, infecting up to 90% of those
exposed. It occurs in epidemics, usually during winter
and spring and involves children who have not previ-
ously been exposed. About 90% of the general adult pop-
ulation have contracted VZV in childhood.
The virus infects the respiratory tract and replicates
for a 2-week incubation period, followed by viremia (vi-
ral dissemination in the bloodstream).
Figure 26-3
Fig. 26-3.
In varicella (chickenpox), fever, malaise,
and headache are followed by the characteristic rash.
The rash of varicella starts on the face and trunk,
spreading to the entire body, including mucous mem-
branes (pharynx, vagina, etc.). The skin vesicles that
form are described as dew on a rose petal: a red base
with a fluid-filled vesicle on top. The fluid becomes
cloudy, the vesicles rupture, and the lesions scab over.
Multiple vesicles arise in patches (crops), and one crop
will form as another crop scabs over. So there are lesions
in different stages! The vesicles will all scab in about 1
week, and the patient then ceases to be contagious.
Zoster (Shingles)
Following a stressed state or lowered cell-mediated
immunity, the latent varicella-zoster virus in the sen-
sory ganglion begins to replicate and migrate to the pe-

ripheral nerves. Burning, painful skin lesions develop
over the area supplied by the sensory nerves. The diag-
nosis of zoster is likely when a patient develops a
painful skin rash that overlays a specific sensory der-
matome
Fig. 26-4. A) 55-year-old male with left, second
trigeminal (V2) nerve involvement; B) 76-year-old fe-
male with left T5-6 involvement.
Since this is the same virus that causes chickenpox,
children and adults who have never contracted varicella
can get chickenpox from exposure to vesicles.
ControllTreatment
A vaccine has been developed for varicella-zoster.
However, since varicella causes only mild disease in
children, there is controversy,over whether a vaccina-
tion program should be instituted.
In adults and immunocompromised patients (with
leukemia or AIDS, for example), the infection can be
more serious, leading to pneumonia and encephalitis. In
these groups zoster immune globulin, which consists of
antibodies against VZV isolated from patients with
zoster, can be given. It will only help if given within days
of exposure (not during rash development). Intravenous
acyclovir, an antiviral drug, appears to decrease the
severity and duration of the infection.
Cytomegalovirus (CMV)
CMV is so-named because infected cells become
swollen (cytomegaly). As with the other herpesviridae,
multinucleated giant cells and intranuclear inclusion
bodies are present.
CMV causes 4 infectious states:
1) Asymptomatic infection: About 80% of adults
in the world have antibodies against CMV. Most of
these infections are asymptomatic.
2) Congenital disease: CMV is one of the
TORCHES (see page 205) organisms that can cross the
placenta and cause congenital disease. CMV is the most
common viral cause of mental retardation. It also
causes microcephaly, deafness, seizures, and multiple
other birth defects. It is thought that this organism will
reactivate during a latent state (as do all the her-
pesviridae). If reactivation occurs during pregnancy,
the fetus may become infected.
3) Cytomegalovirus mononucleosis: CMV causes
a mononucleosis syndrome in young adults similar to
that caused by the Epstein-Barr virus (see Epstein-Barr
virus).
4) CMV can reactivate in the immunocompro-
mised patient (as do all herpesviridae) to cause retini-
CHAPTER 26. HERPESVIRIDAE
206
Figure 26-4
tis (blindness), pneumonia, disseminated infection, and
even death.
Interestingly, CMV causes different diseases in 2 dif
ferent immunocompromised populations: patients with
AIDS versus patients who have undergone bone mar-
row transplantation. In AIDS patients, as the CD4 T-
lymphocyte count drops below 50-100 cells per cc of
blood, they frequently develop CMV viremia (CMV in
the blood), CMV retinitis (leading to blindness unless
treated), and CMV colitis (causing diarrhea). AIDS pa-
tients infected with CMV rarely develop pneumonia. In
contrast, bone marrow transplant patients who are
CMV antibody positive (representing prior infection
and risk of reactivation) or receive a CMV positive donor
bone marrow are at high risk of developing CMV pneu-

Figure 26-5
monitis. CMV pneumonitis is a severe pneumonia, often
leading to death in this population. The transplant pa-
tients can also develop CMV viremia and colitis but do
not develop retinitis.
Marrow Transplant = CMV pneumonitis
AIDS = CMV retinitis
Fig. 26-5.
When you work in the hospital, you will fre-
quently send special blood cultures for CMV from febrile
organ transplant patients (on immunosuppressive
drugs that prevent organ rejection), AIDS patients, and
even children who have leukemia or lymphoma. The
CMV virus invades the white blood cells (see Epstein-
Barr virus below) and so there is a higher yield if the
buffy coat (layer of white cells in centrifuged blood) is
cultured.
Epstein-Barr Virus (EBV)
EBV, another of the herpesviridae, causes the famous
disease mononucleosis and is involved in certain can-
cers such as Burkitt's lymphoma.
A general concept that helps us understand the way
EBV is involved in these disease processes is transfor-
mation.
Transformation and Malignant Potential
In mononucleosis, EBV infects the human B-cells.
EBV actually binds to the complement (C3d) receptor on
cells. Once internalized, EBV will change the infected
cell so that the cell does not follow normal growth con-
trols. These changed or transformed cells proliferate
and pass on copies of the EBV DNA to their progeny.
The EBV DNA remains in the latent state as multiple
CHAPTER 26. HERPESVIRIDAE
20 7
copies of circular DNA. In some of the cells, the EBV ac-
tivates and proliferates, and cell lysis with viral release
occurs.
Interestingly, the transformed cells, which up to this
point are acting as malignant (cancer) cells, suddenly
disappear, with resolution of the mononucleosis illness.
It is thought that the immune system destroys the in-
fecting virus as well as the abnormal B-cells.
EBV has been found in the cancer cells of Burkitt's
lymphoma, a B-cell lymphoma affecting children in
central Africa. Since this cancer does not develop in
other areas of the world where EBV infection occurs, it
is thought that EBV may be just a co-factor in the ma-
lignant transformation. It has been discovered that all
Burkitt's lymphoma cells carry a chromosomal arm
translocation. This translocation activates a chromoso-
mal oncogene. The infection of B-cells by EBV results
in rapid uncontrolled B-cell growth, which may in-
crease the frequency of this key and deadly chromoso-
mal arm translocation (see more about oncogenes in
Chapter 25).
Latent EBV infections in immunosuppressed pa-
tients can reactivate, resulting in transformation and
uncontrolled growth of the B-cell line. Lymphoma and
other lymphoproliferative diseases in these patients
may be secondary to this EBV reactivation.
Mononucleosis
"Mono" is a disease of young adults. As with many vi-
ral infections, the lower the socioeconomic class, the ear-
lier children are infected and the milder the disease (for
example: chickenpox and polio). American teenagers,
living in a higher socioeconomic class with better sani-
tation, handwashing, etc., are infected later in life
through social contact, usually kissing. Thus the refer-
ences to "kissing disease."
Patients with mononucleosis develop fever, chills,
sweats, headache, and a very painful pharyngitis. Most
will have enlarged lymph nodes (as the B-cells multiply)
and even enlarged spleens. Blood work reveals a high
white blood cell count with atypical lymphocytes
seen on the blood smear. These are large activated T-
lymphocytes. The blood also has heterophile anti-
body, which is an antibody against EBV that cross
reacts with and agglutinates sheep red blood cells. This
can be used as a rapid screening test for mononucleosis
(Monospot test).
HHV 8
HHV 8 is a newly identified herpesvirus that appears
to cause Kaposi's sarcoma! (See "Malignancy" subhead-
ing in Chapter 25 for further information.)
Fig. 26-6.
Summary of the herpesviridae.
Figure 26-6 HERPESVIRIDAE
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple' ,MedMaster

CHAPTER 27. THE REST OF THE DNA VIRUSES
CHAPTER 27. THE REST OF THE DNA VIRUSES
Figure 27-1
We have covered in detail the two H's of the HHAPPPy
DNA viruses: herpesviridae and hepatitisviridae. We
will now briefly cover the poxviridae, papovaviridae,
adenoviridae, and parvoviridae.
POXVIRIDAE
Fig. 27-1.
Poxviridae is structurally the most com-
plex of all known viruses. It is a brick-shaped box, POX
in a box, and has at its center a large complex DNA
genome coding for hundreds of proteins. The DNA is or-
ganized into a dumbbell shape with structural proteins,
surrounded by two envelopes. This virus carries many
outs own enzymes and, unlike other DNA viruses, repli-
cates in the cytoplasm.
Smallpox
Poxviridae do NOT cause chickenpox. Chickenpox is
caused by the herpesviridae varicella-zoster. This big
complex pox-box is no chicken! Poxviridae used to cause
smallpox.
Why do we say used to cause smallpox? Answer: The
last case of smallpox was in 1977 and it is thought that
this virus has been eradicated from the planet earth!!!
Pox has been placed in a box and buried.
For more than 3 thousand years this highly conta-
gious virus spread via the respiratory tract, causing pox
skin lesions and death. A concerted vaccination and sur-
veillance program conducted by the World Health Or-
ganization brought this tyranny to an end.
The Vaccine and Why It Worked
1) A vaccine was developed that induced solid, last-
i ng immunity. The vaccine contained vaccinia virus, an
avirulent form of poxviridae, which induced immunity
to virulent poxviridae.
2) Smallpox only infected humans. There are no ani-
mal reservoirs that can harbor this virus and protect it.
209
3) All poxviridae infections produced clinically overt
smallpox. Every smallpox attack was obvious, so mem-
bers of the World Health Organization could localize
communities that needed vaccination. The virus could
not hide as an asymptomatic infection or in a latent
state.
Molluscum Contagiosum
Fig. 27-2.
You mole! You scum! molluscum. A pox
virus causes these small, 1-2 mm in diameter, white
bumps that have a central dimple (seen to the right of
the mole in this figure). They are similar to warts with
benign hyperproliferation of epithelial cells. You will
probably first see these lesions on AIDS patients, who
frequently develop them.
PAPOVAVIRIDAE
There are 3 members of the PA-PO-VA VIRIDAE:
PA: PApilloma virus causes human warts and cervical
cancer.
PO: POlyomavirus has 2 members: human BK and JC
virus.
VA: Simian VAcuolating virus does not infect humans.
PAPOVAVIRIDAE:
With these viruses think O
O for circular double-stranded DNA (naked icosahedral
capsid)
O for round warts
O for round cervix
Papilloma Virus
Different strains of the papilloma virus can cause
warts and cervical cancer.

Figure 27-2
Warts
There are many strains of papilloma viruses. They
have a tropism for squamous epithelial cells, and differ-
ent strains like certain anatomic regions: common
warts, genital warts, laryngeal warts. Warts are benign
hyperproliferations of the keratinized squamous ep-
ithelium. Most will resolve spontaneously within 1-2
years. For unclear reasons many people do not develop
warts despite the ubiquitous nature of the papilloma
virus. Perhaps in these unaffected individuals the virus
remains latent or is effectively controlled by the host im-
mune system.
Cervical Cancer
Cervical dysplasia and carcinoma are associated with
sexual activity and previous exposure to certain strains
(type 16 and 18) of human papilloma virus.
Think of PAPilloma virus and the PAP smear, which
is used to detect early dysplastic cellular changes. The
Pap test has resulted in early detection of cervical dys-
plastic changes and has significantly reduced the pro-
gression to cervical cancer.
Polyomavirus
Two polyoma viruses infect humans. They were
named after the initials of the patient from whom the
virus was discovered. Both are ubiquitous and infect
worldwide at an early age.
CHAPTER 27. THE REST OF THE DNA VIRUSES
210
BK Polyomavirus
1) As ubiquitous as the Burger King at every high-
way exit.
2) Causes mild or asymptomatic infection in chil-
dren.
JC Polyomavirus
Fig. 27-3. JC Polyomavirus is similar to BK but also
causes an opportunistic infection in immunocompro-
mised
patients
called
Progressive
Multifocal
Leukoencephalopathy (PML). In this disease, pa-
tients develop central nervous system, white matter
damage. Visualize shoppers at J.C. Penney with PML,
walking around the store with memory loss, poor
speech, and incoordination.
ADENOVIRIDAE
The ADENoviridae cause upper respiratory tract in-
fections in children. Visualize A DEN full of coughing,
sneezing children. Studies estimate more than 10/0 of
childhood respiratory infections are caused by strains of
adenoviridae, and virtually all adults have serologic ev-
idence of prior exposure. Infection can result in rhinitis,
conjunctivitis, sore throat, and cough. This can some-
times progress to lower respiratory tract pneumonia in
children.
Viral respiratory illness in children in order of fre-
quency:

CHAPTER 27. THE REST OF THE DNA VIRUSES
Figure 27-4
21 1

PARVOVIRIDAE
Fig. 27-4.
PARvovirus is the smallest icosahedral
virus and has a single strand of DNA. Simple as a Par-
ONE golf course!
212
CHAPTER
27.
THE REST OF THE DNA VIRUSES
#1
RSV
It causes a childhood disease called erythema in.
#2
Parainfluenza
fectiosum ( Fifth disease), characterized by fever and a
#3
Rhinovirus
"slapped face" rash on the cheeks.
#4
Adenoviridae
Fig. 27-5.
Summary of the Rest of the DNA Viruses.
Figure 27-5 THE REST OF THE DNA VIRUSES
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
©MedMaster

CHAPTER 28. THE REST OF THE: RNA VIRUSES
We have covered in previous chapters some of the RNA
viruses: retroviridae, orthomvxoviridae. and paramyx-
oviridae. We will now briefly review:
1I The ARthropod Borne viruses, which are called
arboviruses: These RNA viruses include the to-
gaviridae, flaviviridae, and bunyaviridae.
Although not transmitted by arthropods. rubivirus
which causes rubella) and hantavirus (hantavirus
pulmonary syndrome) will be discussed here because
they are members of the togaviridae and bunyaviridae.
respectively.
2) The picornaviridae, which are a large group
of enteroviruses (ENTERO=Gl, fecal-oral transmis-
sion): hepatitis A virus; poliovirus; coxsackie A. B; and
echovirus.
3) Viruses that cause the common cold: rhinovirus
(really in the picornaviridae family) and coronaviridae.
4) Viruses that cause diarrhea: rotavirus, and call
civiridae (which includes the Norwalk virus).
5) Rabies caused by the rhabdoviridae.
THE ARBOVIRUSES
Fig. 28-1. The arboviruses include the bunyaviri-
dae, togaviridae, and flaviviridae. All are
transmitted by blood-sucking arthropods and cause
fever and encephalitis. The legendary U.S. logger Paul
Bunyan, wearing a toga, has a rich flavor that
attracts mosqui- tos and other arthropods. lie is about
to chop down a tree (ARBOL in Spanish). You can
well imagine Paul Bunyan has quite a headache
(encephalitis) with that blood-sucker clinging to his
toga!
Togaviridae
Two members of this family infect humans:
1) Alpha viruses are mosquito-borne and cause
enchephalitis, an inflammation of the brain with fever.
headache. altered levels of consciousness. and focal neu-
rologic deficits.
2) Rubivirus causes rubella.
Alpha Viruses
The 3 main alpha viruses that cause encephalitis all
infect horses. birds, and humans. They use the mosquito as
a vector.
Fig. 28.2. The togaviridae alpha viruses. Picture Paul
Bunyan riding on a roller-coaster wearing his toga ( to-
gaviridae), with a mosquito (mosquito vector) on his
head. The other passengers scream the names of the 3
main diseases caused by the togaviridae alpha viruses,
which are named by geographic region:
Figure 28-1
WEE: Western equine encephalitis (western U.S.
MA Canada).
EEE: Eastern equine encephalitis (eastern U.S.)
.
VEE: Venezuelan equine encephalitis (South and (cen
tral America. southern U.S.).
Rubivirus
Rubivirus is a togavirus, but it is not an arbovirus be
cause humans are the only infected creatures. Ru-
bivirus causes rubella, which is a mild febrile illness
with a rash. The importance of this virus lies in its ability
to cross the placenta and cause terrible congenital
defects, especially in the first trimester.
Rubella ("German measles") is a mild measles-like
illness. Like measles, rubella is contracted by respiratory
secretions and has a prodrome of fever and flu
symptoms This is followed by a red maculopapular rash
that sprees from forehead to face to torso to extremities.
Unlike measles, patients are leas "sick," complications such
as en-cephalitis do not occur, and the rash lasts only 3 days,
oat 6. Thus its other name: "3-day measles. "Young
women can develop self-limiting arthritis with the
infection.
The R in TORCHES see Fig. 26.21 stands for the
feared congenital rubella. The risk of rubella-induced
214
Figure 28-2
congenital defects is greatest early in fetal development
Body areas affected in congenital rubella include:
when cell differentiation is at a peak. Rubivirus-infected
human embryo cells demonstrate chromosomal break-
1) Heart: patent ductus, interventricular septal de-
age and inhibition of mitosis.
fects, pulmonary artery stenosis, others.
CHAPTER 28. THE REST OF THE RNA VIRUSES
215

2) Eye: cataracts, chorioretinitis, others.
3) CNS: mental retardation, microcephaly, deafness.
A live attenuated rubella vaccine is given to all young
children in the U. S. It is not recommended for pregnant
women because of the theoretical risk of fetal infection.
However, there is no evidence that this vaccine causes
congenital defects.
Pregnant women are routinely screened for immunity
to rubella. If they do not have antibody to rubivirus,
they will receive immunization after delivery.
Flaviviridae
The flaviviridae share many similarities with the to-
gaviridae:
The morphology is similar (see Fig. 28-10).
They cause encephalitis, with names based on geo-
graphic location (Japanese encephalitis, Russian en-
cephalitis, etc.).
Fig. 28-3.
The flaviviridae are spread by a mos-
quito vector, infecting humans and birds.
The flaviviridae also cause the febrile diseases yel-
low fever and Dengue fever.
1) Yellow fever was made famous by the Panama
Canal project. This flavivirus (yellow fever virus) was
transmitted to canal workers by mosquitoes. One week
later they would develop hepatitis with jaundice (yellow
appearance), fever, backache, nausea, and vomiting.
Once the vector was found to be a mosquito, insecti-
cides were used to control the disease. Spraying contin-
ues in the southern U. S. and Latin American urban
centers, virtually eliminating urban yellow fever.
2) Dengue fever is a mosquito-borne febrile disease
that occurs in the tropics (Puerto Rico, Virgin Islands).
It is also called break-bone fever because of the painful
backache, muscle and joint pain, and severe headache.
Painful fever!
There is a new severe variant called Dengue hem-
orrhagic fever, which causes hemorrhage or shock in
children with a mortality rate of almost 10%.
Figure 28-3
CHAPTER 28. THE REST OF THE RNA VIRUSES
21 6
West Nile Virus is a flavivirus spread by mosquitoes
(which feed on infected birds) or blood transfusion. It has
been found in Africa, East Europe, West Asia, Middle
East and increasingly more frequently in the U.S. Most
cases present as a mild flu-like illness, but may present
with encephalitis and death, particularly in the elderly.
Bunyaviridae
Bunyaviridae also cause diseases characterized by
fever and encephalitis, such as California encephali-
tis and Rift Valley fever. For comparison of the bun-
yaviridae with the other arboviruses (toga and flavi),
see Fig. 28-10.
Hantavirus Pulmonary Syndrome
In May 1993 reports began to emerge from the Four
Corners area of New Mexico, Arizona, Colorado, and
Utah, of an influenza-like illness followed by sudden res-
piratory failure, frequently culminating in death. Many
of these patients were previously healthy adults. The
etiologic agent is a virus in the bunyaviridae family in the
genus hantavirus. Hantavirus had previously only been
associated with the disease hemorrhagic fever with
renal failure seen in Asia and Europe. The deer mouse
is the reservoir for this virus and exposure to the drop-
pings of these rodents accounts for human infections.
More than 50 cases of what is now being called han.
tavirus pulmonary syndrome have been confirmed
in 14 states. Patients typically present with high fevers,
muscle aches, cough, nausea, and vomiting. Their heart
and respiratory rate is rapid and blood work may reveal
a high white blood cell count and a high red blood cell
count. The lung capillary permeability is disrupted re-
sulting in fluid leakage into the alveoli (pulmonary
edema). The fluid-filled alveoli are unable to deliver
oxygen to the bloodstream, and intubation with me-
chanical ventilation is required to enhance oxygenation
in close to 90% of patients. Death follows in 80% of pa-
tients within a median of 9 days.
This illness should be considered in young adults
with influenza-like symptoms who develop pulmonary
edema. Investigational treatment with ribavirin is
currently recommended.
(MMWR, 1994; Duchin, 1994).
PICORNAVIRIDAE
This family of viruses all have similar structure and
replication.
There are 2 genera: enterovirus and rhinovirus.
Enterovirus
1) Enteroviruses have 5 subgroups:
a) Poliovirus
b) Coxsackie viruses A and B

c) Echovirus
d) New enteroviruses
e) Hepatitis A
These are all called enteroviruses because they infect
intestinal epithelial and lymphoid (tonsils, Peyer's
patches) cells. They are excreted in the feces and spread
by the fecal-oral route. The replication in the tonsils also
results in viral shedding from pharyngeal secretions.
Poliovirus will be discussed first as it causes the im-
portant paralytic disease, poliomyelitis. Hepatitis A is
covered in Chapter 24. The remainder will be discussed
together as there is significant overlap in the diseases
they cause.
2) Rhinovirus causes the common cold and will be
discussed last.
Poliovirus
Poliovirus has the ability to infect cells in the:
1) Peyer's patches of the intestine.
2) Motor neurons.
This tropism explains:
1) The fecal-oral mode of transmission.
2) The disease paralytic poliomyelitis.
Polio was one of the feared diseases of the 20th cen-
tury. In the 1950's 6 thousand cases of paralytic polio oc-
curred each year in the U. S.
This disease was in part due to improvements
in sanitation. Children tend to have fewer paralytic
complications with poliovirus infection than do adults.
As sanitation improved and fecally contaminated sub-
stances were cleared, fewer people were exposed to po-
liovirus as children, and thus more adults were infected.
The chances of developing paralytic poliomyelitis in-
creases as one gets older, thus explaining the increase in
paralytic poliomyelitis with improvements in sanitation.
Now that a vaccine has been developed, the incidence
has markedly diminished.
The Disease Polio
The virus initially replicates in the tonsils and Peyer's
patches, spreading to the blood, and across the blood-
CNS barrier to the anterior horn of the spinal cord.
Because of the initial replication in the tonsils, the
virus can be spread by respiratory secretions, as well as
the usual fecal-oral route, early in the course of infection.
There are 3 disease manifestations:
1) Mild illness: An asymptomatic infection or a mild
febrile viral illness is the most common form. This es-
pecially occurs in infants in less-developed nations,
where the sanitation is poor.
2) Aseptic meningitis: Fever and meningismus can
develop as the poliovirus infects the meninges. Recovery
is complete in 1 week.
CHAPTER 28. THE REST OF THE RNA VIRUSES
21 7
3) Paralytic poliomyelitis: This is the feared man-
ifestation of poliovirus infection!!! The viral infection de-
stroys presynaptic motor neurons in the anterior horn of
the spinal cord as well as the postsynaptic neurons leav-
ing the horn. The damage to the exiting motor neurons
results in clinical manifestations of peripheral motor
neuron deficits, while the presynaptic neuron damage
causes central motor neuron deficits.
This disease is truly terrifying. A mild febrile illness
resolves, 5 to 10 days later the fever recurs, followed by
meningismus and then flaccid asymmetric paralysis.
The paralytic disease can range from 1 leg or arm to
paraplegia, quadriplegia, and even respiratory muscle
dysfunction. The later more serious events usually oc-
cur in persons older than 15 years.
The affected extremities early in the course will have
painful muscle spasms. Asymmetric muscle paralysis
develops. Ultimately, atrophy and loss of reflexes occur
(there are no sensory losses).
Vaccines
The inactivated polio vaccine, developed by Jonas
Salk, contains formalin-killed viruses that are in-
jected subcutaneously, provoking an IgG antibody re-
sponse that will protect against future viremia. This is
used in Scandinavia today with excellent results.
The oral polio vaccine was developed by Albert B.
Sabin and is currently used in the U. S. This vaccine
contains attenuated poliovirus that has lost the abil-
ity to multiply in the CNS. It is taken orally and repli-
cates and sheds in the feces in the normal fashion but
does not cause paralytic poliomyelitis.
This vaccine works by supplanting the wild type
(disease-causing) poliovirus with a docile attenuated
counterpart. We are NOT eliminating poliovirus com-
pletely but are only trying to eliminate the virulent
strain. In the case of the smallpox vaccine, immuniza-
tion of the population depleted the viral reservoir (hu-
mans), and the virus was completely eliminated.
There are good and bad things about the oral Sabin
vaccine, and there has been some debate stemming from
lawsuits and comparisons between our system and that
of Scandinavia.
Positives
1) It is an oral vaccine.
2) The oral route and full replication allow formation
of both IgG in the blood and secretory IgA in the GI tract.
3) The attenuated virus is spread to contacts, re-
sulting in a secondary infection and immunity in these
individuals.
Negatives
Vaccine-associated paralytic poliomyelitis: The
vaccine can pick up virulence and cause paralysis in the

person taking the vaccine or in those exposed to shed-
ding (parents changing a vaccinated infant's diapers).
This is very rare (1/2.6 million doses), but out of the 138
cases of paralytic polio between 1973 and 1984, 105
cases were considered vaccine related. Persons with im-
munodeficiency states should not receive the oral at-
tenuated vaccine.
Ultimately, both vaccines have been very effective,
resulting in almost complete control of the virulent po-
liovirus in vaccinated geographic regions.
Coxsackie A and B, Echoviruses, New
Enteroviruses
The remainder of the Enteroviruses in the Picor-
naviridae family are responsible for a variety of dis-
eases. There is overlap since the different viruses can
cause the same clinical symptoms.
The Coxsackie viruses (A and B), the echoviruses, and
the new enteroviruses all have multiple serotypes and
all can cause:
1) Asymptomatic or mild febrile infections.
2) Respiratory symptoms ("cold").
3) Rashes.
4) Aseptic meningitis: The enteroviruses are the
most common cause of non-bacterial (aseptic) meningi-
tis in the U. S.
Coxsackie A
Coxsackie A can be differentiated from B by its effect
on mice. Coxsackie A causes paralysis and death of the
mouse with extensive skeletal muscle necrosis. Cox-
sackie A also causes:
Herpangina. A mild self-limiting illness character-
ized by fever, sore throat, and small red-based vesicles
over the back of the throat.
Coxsackie B
Coxsackie B causes less severe infection in mice but
multiple organs can be damaged, such as heart, brain,
liver, pancreas, and skeletal muscle. It also causes:
CHAPTER 28. THE REST OF THE RNA VIRUSES
Figure 28-4 ILLNESSES CAUSED BY ENTEROVIRUSES
21 8
1) Pleurodynia. Fever, headache, and severe lower
thoracic pain on breathing (pleuritic pain) mark the Cox-
sackie B virus respiratory infection.
2) MyocarditisIPericarditis. Infection and inflam-
mation of the heart muscle and pericardial membrane
can result in self-limited chest pain or more serious ar-
rhythmias, cardiomyopathy, and heart failure. Many
viruses can cause this, but Coxsackie B is associated
with 50% of the cases!
Fig. 28-4.
Comparisons of the enteroviruses.
VIRUSES THAT CAUSE THE "COLD"
Fig. 28-5.
Rhino with the common cold, drinking a
Corona beer. This will help you remember that the rhi-
novirus and the coronaviridae both cause the com-
mon cold.
More than 100 different serotypes of rhinovirus are re-
sponsible for the "common cold." Transmission occurs by
hand-to-hand spread of mucous membrane secretions.
The coronaviridae cause a cold indistinguishable
from the rhinovirus common cold. About 15% of adult
common colds are caused by the coronaviridae.
VIRUSES THAT CAUSE DIARRHEA
Viruses that cause gastroenteritis are acquired by the
fecal-oral route and usually prey on infants and young
children, although outbreaks among adults do occur.
Fever, vomiting, abdominal pain, and diarrhea follow a
1-2 day incubation, and symptoms resolve within 4-7 days.
Infants die secondary to loss of fluids and electrolytes.
DIARRHEA = DEATH BY DEHYDRATION
Two groups of viruses have been implicated in diar-
rhea: calciviridae (including the Norwalk virus) and ro-
tavirus.
Fig. 28-6.
If your calico cat develops diarrhea, ro-
tate the kitty litter frequently, or rotate the calico
cat off to Norway. This picture will help you remem-
ber that viral gastroenteritis (diarrhea) is caused by
DISEASES
Coxsackie
ECHOvirus &
A
B
New enterovirus
Asymptomatic infections
+
+
+
Respiratory infections
+
+
Rashes ("exanthems")
+
+
+
Aseptic meningitis
+
+
+
Herpangina
+
-
-
Pleurodynia
-
+
-
Myocarditis
-
+
-
Pericarditis
-
+
+
Figure 28-5
caliciviridae, including the Norwalk virus, and the
very common rotavirus.
1) Caliciviridae primarily infects young children
and infants. The gastroenteritis is indistinguishable
Figure 28-6
CHAPTER 28. THE REST OF THE RNA VIRUSES
from that of rotavirus, including diarrhea, vomiting,
and fever.
2) Norwalk virus can occur in adults, but the virus
is named after an outbreak in a Norwalk, Ohio, ele-
mentary school. In that episode, 50% of students devel-
oped diarrhea and severe vomiting.
3) Rotavirus is a member of the family reovirus
(Respiratory Enteric Orphan). It is the number one
cause of acute infectious diarrhea and a major
cause of infant mortality worldwide. More than 1 mil-
lion infant deaths per year are secondary to rotavirus.
Note: Only bacterial cholera causes worse dehydra-
tion than the rotavirus.
Treatment for all of these is supportive, with IV flu-
ids and electrolytes. Oral rehydration therapy has
revolutionized the treatment of diarrhea in underdevel-
oped nations, where IV fluids are a rare commodity.
RHABDOVIRIDAE AND RABIES
Fig. 28-7.
Rhabdoviruses have bullet-shaped, en-
veloped, helical symmetry nucleocapsids.
Rabies virus is the only virus in this family that nor-
mally infects humans. The rabies virus can infect all
warm-blooded animals, with dogs, cats, skunks, coyotes,
foxes, raccoons, and bats serving as reservoirs. The in-
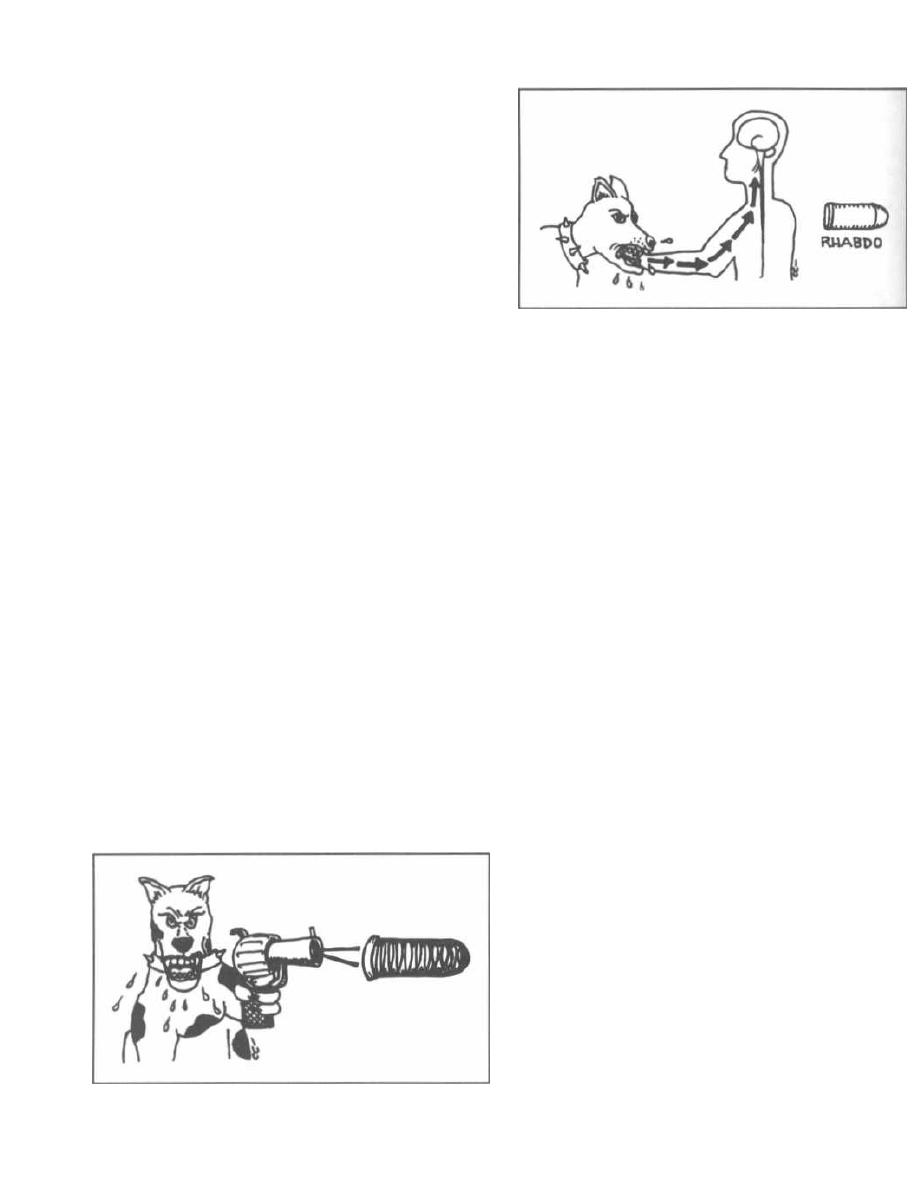
fected creatures develop encephalitis and can become fear-
less, aggressive, and disoriented. The famous stories of the
mad farm dog or the wild wolf that stumbles fearlessly into
an Alaskan town have popularized this conception.
Fig. 28-8.
When a human is bitten, the virus repli-
cates locally at the wound site for a few days, then mi-
grates (slowly over weeks to a year) up nerve axons to the
central nervous system, causing a fatal encephalitis.
Fig. 28-9.
Brain cells in rabies demonstrate neuro-
pathic changes and pathognomonic collections of virions
in the cytoplasm called Negri bodies.
Following the bite of a rabid animal there is an incu-
bation period that has tremendous variability, rang-
ing from a week to years! Once symptoms develop, there
is rapid progression to death over 1-2 weeks:
1) Prodrome: Infected persons first develop nonspe-
cific symptoms of fever, headache, sore throat, fatigue,
nausea, and painfully sensitive nerves around the
healed wound site. The muscles around the site may
even fasciculate!
2) Acute encephalitis: Hyperactivity and agitation
lead to confusion, meningismus, and even seizures.
Madness!!
3) Classic brainstem encephalitis: The brainstem
infection causes cranial nerve dysfunction and painful
contractions of pharyngeal muscles with swallowing liq-
uids ("hydrophobia"). This results in an inability to
swallow saliva and "foaming of the mouth."
4) Death ultimately occurs secondary to respiratory
center dysfunction. There has only been 1 reported case
of recovery from active rabies.
Through effective control and treatment strategies,
the number of actual cases of rabies has been dramati-
cally reduced.
1) Vaccination of dogs and cats has been very effec-
tive in the U. S., with only 18 cases reported from 1980
to 1993; infection was actually acquired in the U.S. in
only 8 of these cases (Fishbein,1993).
Figure 28-7
CHAPTER 28. THE REST OF THE RNA VIRUSES
220
Figure 28-8
2) When a person has been bitten or an open wound
licked by a possibly rabid animal, the wound should be
aggressively cleaned with soap and water. Washing
alone will significantly lower the risk of infection.
3) The animal should be captured or destroyed. Cap-
tured animals are confined, and if no symptoms develop
in the animal within 10 days, the animal does not have
rabies. If destroyed, the dead animal's brain can be ex-
amined for Negri bodies or tested for uptake of fluores-
cently labeled antibodies to rabies virus.
4) If the animal cannot be captured or the above tests
are positive, the bitten individual should receive human
rabies immune globulin (passive immunization), fol-
lowed by 5 injections of the killed rabies virus vaccine
(active immunization). The idea is to develop immunity
while the virus is still in the prolonged (variable length)
incubation period.
Notice the similarities in the treatment of rabies with
that given to a person presenting with tetanus (see
Chapter 6).
FILOVIRIDAE (Ebola and
Marburg viruses)
EBOLA VIRUS: April 4th, 1995:
It was a summer morning in the city of Kikwit, Zaire
( population 400,000). A hospital laboratory technician
had a fever and splitting headache with aching muscles
and shoulders. He walked out of his home swinging his
arms in loose circles, hoping to shake off the deepening
pain. The feeling remained and a heavy fatigue and
crampy abdominal pain soon settled in. Must be the flu,
he thought. At work, he filled a cup with water and his
hand, now sweaty, trembled as he brought the water to
his lips. His throat hurt as he swallowed. He scarcely no-
ticed the hiccups he suffered for the last half hour as his
gut pain intensified. He stumbled to the rest room and
had a large bowel movement. He staggered up and found
the toilet water blood red. Slumping to the ground, he
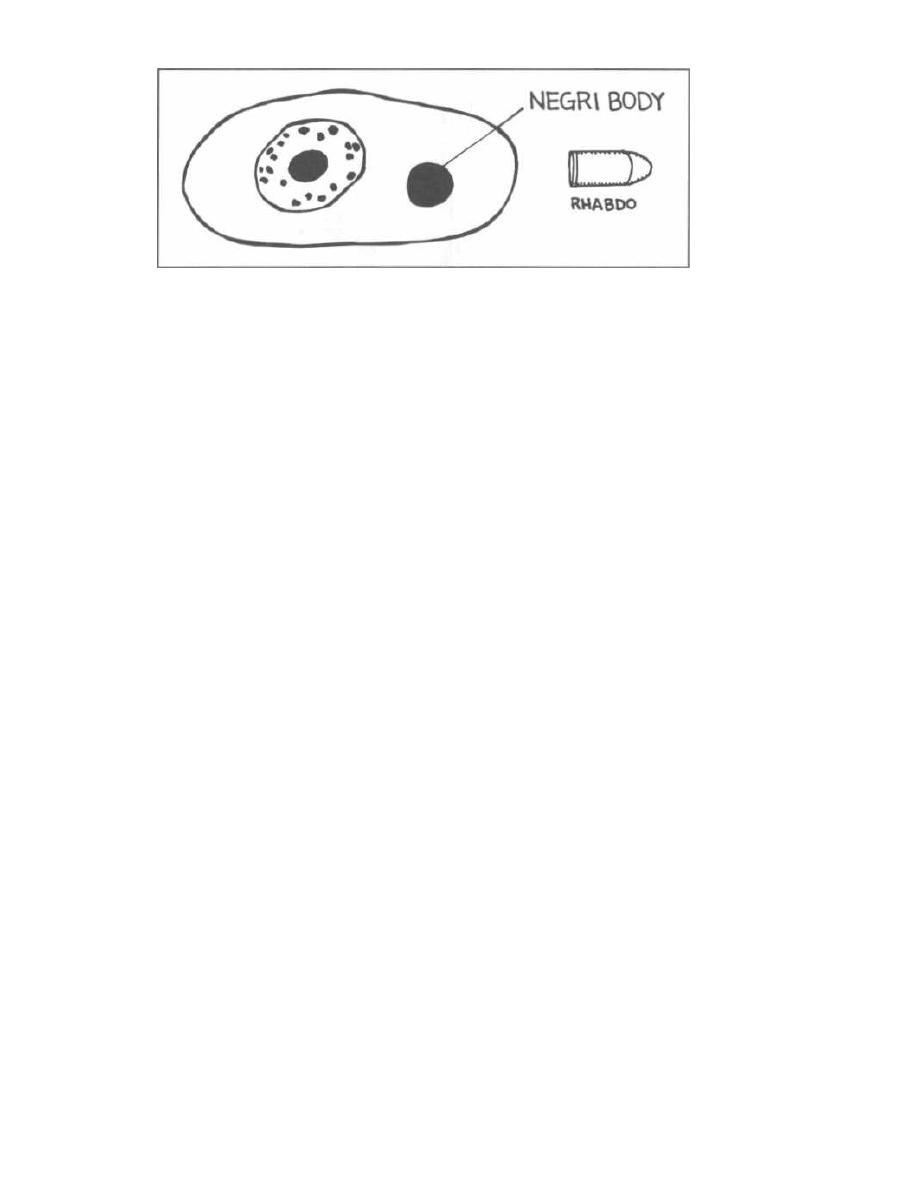
CHAPTER 28. THE REST OF THE RNA VIRUSES
Figure 28-9
coughed and blood began to flow from his nose and
mouth. His pants were soon soiled with bloody diarrhea.
Physicians suspected this patient had a perforated
bowel and took him to surgery. Within 2 weeks of his
presentation, multiple hospital personnel became ill
with similar symptoms: fever (94%), diarrhea (80%),
weakness (74%), dysphagia (41%), hiccups (15%), and
bleeding from mucous membranes--G.I. tract, vagina,
and skin (38%). The disease affected men and women
alike. (MMWR, June 30, 1995). By May 17, 93 persons
were infected and by June 25th, 296 persons.
Suspecting that the deaths were secondary to a viral
hemorrhagic fever (VHF)-like illness, seen in cases of
Ebola and Marburg viruses, blood samples were sent
to the CDC and polymerase chain reaction (PCR) and
enzyme-linked immunosorbent assay (ELISA) tests re-
turned positive for Ebola virus.
Fib, in filoviridae, means "filament" in Latin and de-
scribes the filamentous shape of the RNA viruses Ebola
and Marburg that comprise the filovirus family. They
are responsible for rare outbreaks of viral hemorrhagic
fever in sub-Saharan Africa (Zaire, Sudan, Uganda,
Kenya) or in the U.S. or Europe following contact with
monkeys from these sub-Saharan African areas. Hu-
mans and monkeys are infected during outbreaks but it
is not known what organism serves as reservoir be-
tween epidemics. Serologic studies have demonstrated
a 17% seropositivity for Ebola in selected central
African populations. This is highest in hunter-gatherers
such as the Aka Pygmies (37.5% seropositivity) who
handle freshly killed animals. (Johnson, 1993). It is still
unclear what sets off the infrequent and deadly epi-
demics of viral hemorrhagic fever.
Transmission:
In the Zaire outbreak the most frequently infected
groups were health care workers, home caregivers, and
family members (especially spouses). All were in di-
rect contact with body fluids.
Direct contact with blood, vomitus, urine, stool, or se-
men, from the living or dead patient, appears to be the
most important route of transmission. This likely occurs
via skin or mucous membrane contact with the virus-
22 1
infected body fluids. Reuse of unsterile needles was sig-
nificant in the Kikwit, Zaire epidemic.
Airbourne transmission is an unlikely mechanism in
humans but has been documented in monkeys. Current
CDC guidelines do recommend use of masks and nega-
tive pressure room isolation as there is still some concern
over aerosol transfer during the later stages of illness.
Control and Treatment:
Epidemics have been controlled by barrier precau-
tions to avoid contact with infected body fluids, use of
sterile needles, limiting laboratory blood work, and
proper disposal of corpses (sealed in leakproof material
and cremated or buried in sealed casket). Laundry and
equipment must be incinerated, autoclaved, or washed
with bleach.
There is no known effective anti-viral therapy. Ther-
apy is supportive.
Fig. 28-10. Summary of the "Rest of the RNA Viruses."
References
CDC. Outbreak of Ebola viral hemorrhagic fever - Zaire 1995.
MMWR 1995;44(19)381-382.
CDC. Update: outbreak of Ebola viral hemorrhagic fever -
Zaire, 1995. MMWR 1995;44(25):468-470.
CDC. Update: management of patients with suspected viral
hemorrhagic fever - United States. MMWR 1995;44(25):
475-479.
Duchin JS, et al. Hantavirus pulmonary syndrome: a clinical
description of 17 patients with a newly recognized disease.
N Engl J Med 1994;330:949-955.
Fishbein DB, Robinson LE. Current Concepts: Rabies. N Engl
J Med 1993; 329:1632-1638.
Hantavirus pulmonary syndrome-United States, 1993.
MMWR Morb Mortal Wkly Rep 1994;43:45-48.
Johnson ED, Gonzalez JP, Georges A. Filovirus activity
among selected ethnic groups inhabiting the tropical forest
of equatorial Africa. Transactions of the Royal Society of
Tropical Medicine and Hygiene 1993;87:536-538.
Peters CJ. Marburg and Ebola virus hemorrhagic fevers. In:
Mandell GL, Bennett JE, Dolin R, eds. Principles and Prac-
tice of Infectious Diseases; 4th edition. New York: Churchill
Livingstone, 1995;1543-1546.
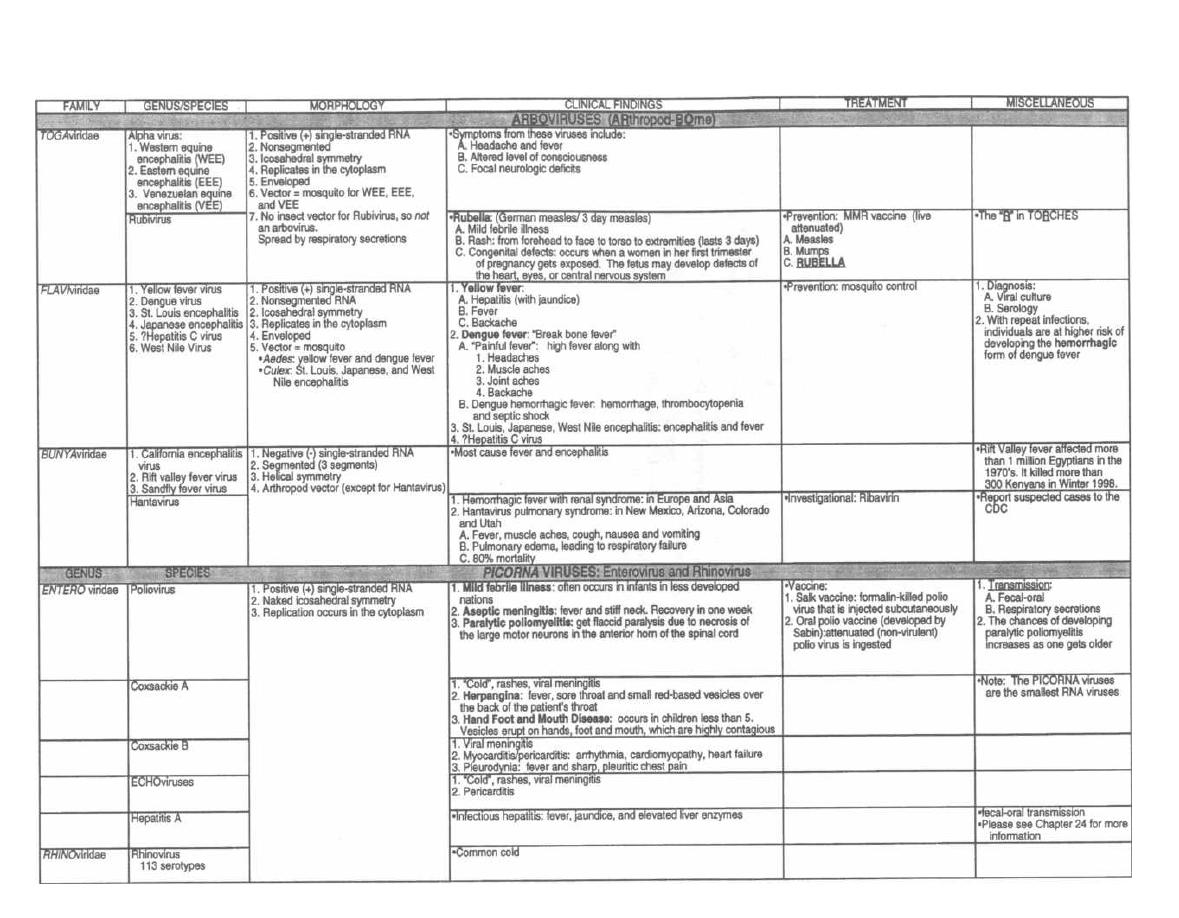
Figure 28-10 THE REST OF THE RNA VIRUSES
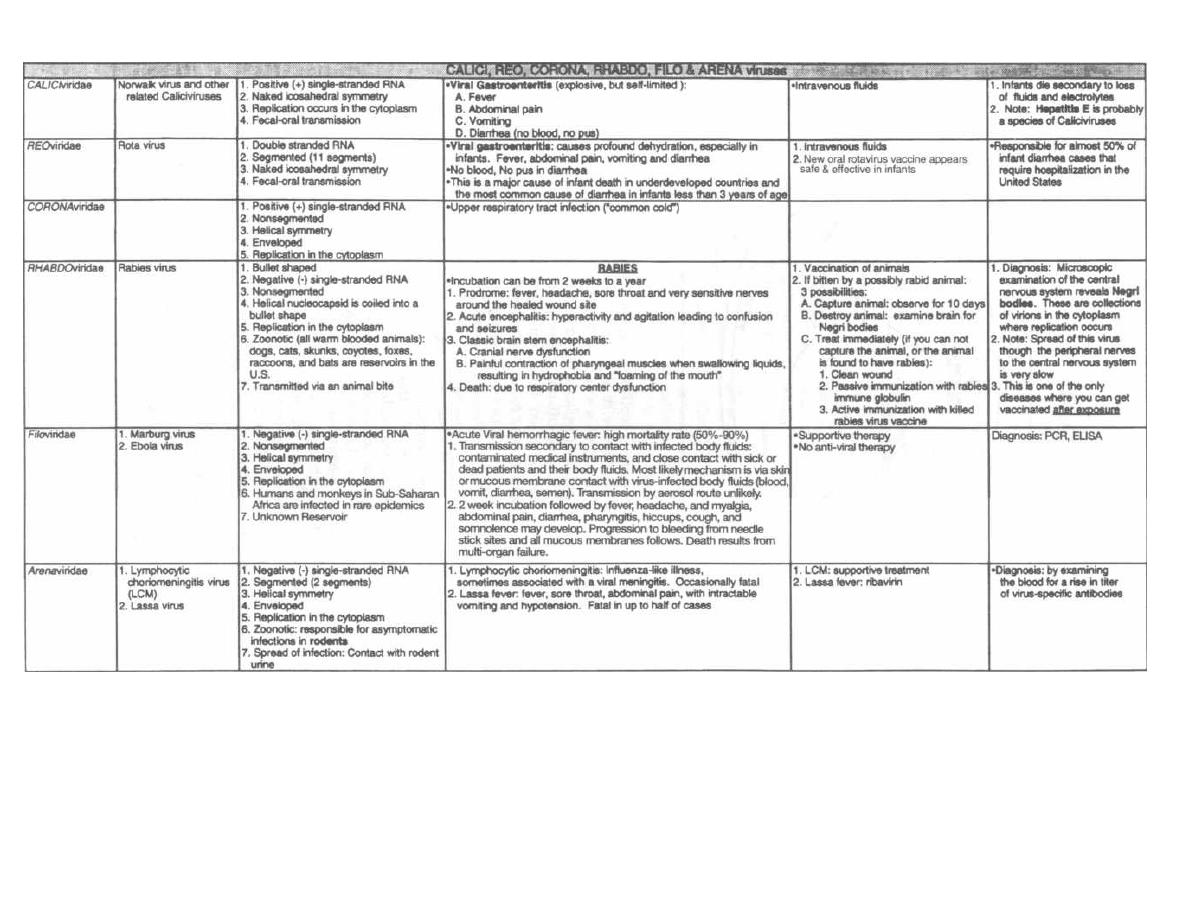
Figure 28-10 (continued)
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
CMAedMaster
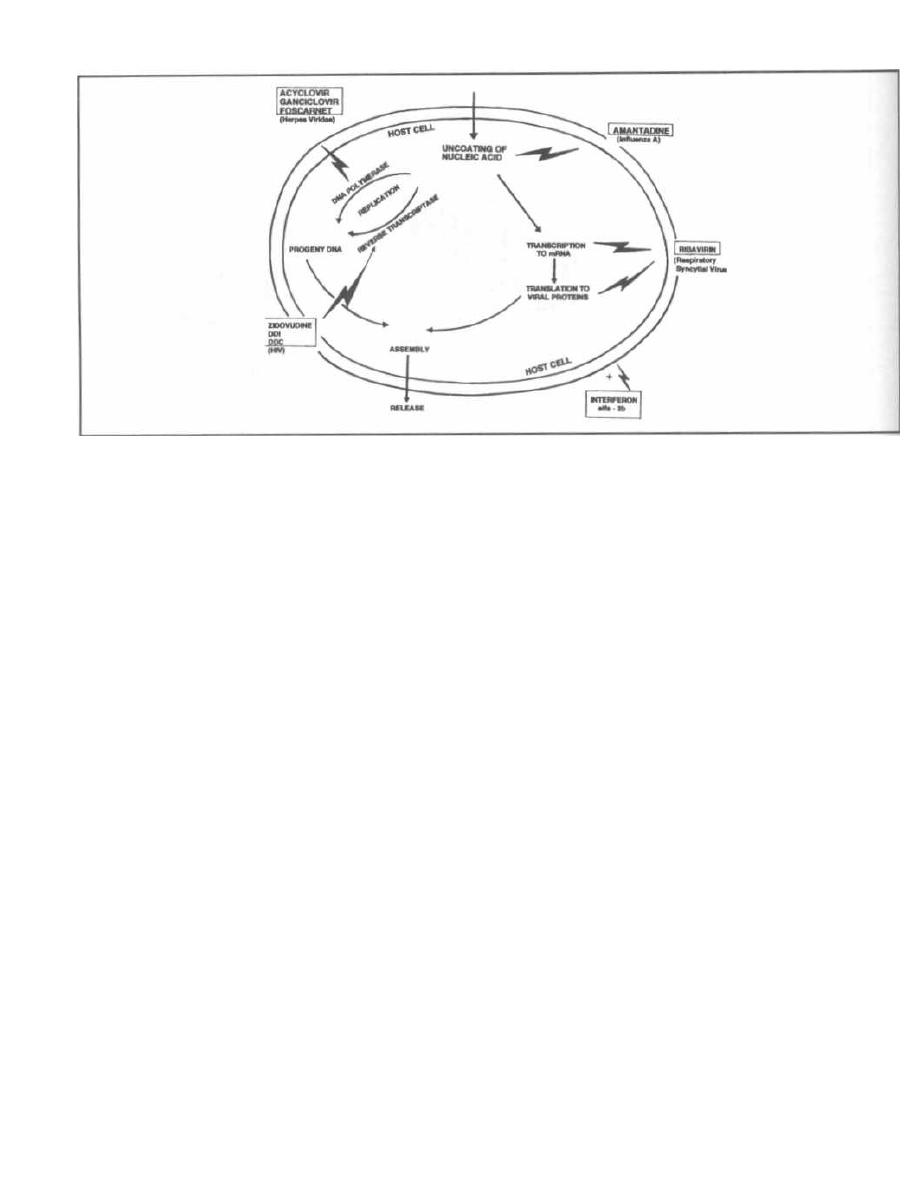
CHAPTER 29. ANTI-VIRAL MEDICATIONS
Figure 29-1
The virus is a tough creature to kill. It has NO peptido-
glycan wall, NO ribosomes, and NO cell membrane. All it
has is a protein coat, nucleic acid strand, and a few sim-
ple enzymes. The only thing these critters do is replicate
and then hang out in a latent state. The current antiviral
agents attack steps in viral replication much like the
chemotherapeutic agents attack replicating tumor cells.
Fig. 29-1. Site of action of antiviral drugs.
Two important concepts:
1) These drugs attack steps in actively replicating
viruses and have no effect on latent viruses. For this
reason these drugs are only viirustatic (not virucidal).
2) Most of these drugs are nucleotide analogues.
They look just like the viral nucleotides but do not func-
tion appropriately. As such, they are taken up and used
by viral DNA polymerase or reverse transcriptase and
are like monkey wrenches thrown into the gears of repli-
cation. They inhibit the DNA polymerase or reverse
transcriptase and are also incorporated into the grow-
ing DNA strand, resulting in chain termination.
ANTI-HERPESVIRIDAE DRUGS
Both acycloviir and ganciclovir are guanine ana-
logues that act against the herpes family. There is a key
difference between them. To become active, acyclovir
must first be phosphorylated by a virus-specific thymi-
dine kinase. Most of the herpesviridae have this enzyme
while human cells do not. For this reason acyclovir is ac-
224
tive only against herpesviridae and has limited toxicity
to our cells. One of the herpesviridae, cytomegalovirus
(CMV), lacks thymidine kinase and so acyclovir is less
active for CMV infections. On the other hand, ganciclovir
is not dependent on a virus-specific thymidine kinase
for phosphorylation. It kills ALL the herpesviridae in-
cluding CMV. It is also toxic to some rapidly replicating
human cells such as neutrophils and platelets (causes
neutropenia and thrombocytopenia).
Acyclovir
("A cycle")
Fig. 29-2. To remember that ACYCLovir is used
to treat infections caused by the herpes family, visual-
ize A CYCLE traversing the heights of a huge herpes
cold sore.
Clinical Uses
Studies demonstrate that if acyclovir is given very
early, it reduces the severity and duration of all herpes
simplex and varicella-zoster (V-Z) infections, such as
cold sores (mucocutaneous herpes simplex infections),
varicella (chickenpox), and zoster (shingles). However,
because these infections are self-limiting and mild, acy-
clovir is currently not recommended for these diseases.
In the immunocompetent host, it is reserved for more
serious infections such as herpes simplex encephalitis
and herpes simplex and zoster infections of the eye. It is
also approved for herpes simplex genital infections.
Figure 29-2
In the immunocompromised host, it is used for most
herpes infections (mucocutaneous, varicella, and zoster).
Acyclovir is not used for CMV or Epstein-Barr virus
infections.
Fig. 29.3. Adverse effects are minimal. With high in-
travenous doses acyclovir (A CYCLE) may crystallize in
the renal tubules, resulting in reversible renal toxicity.
About 19 of patients have CNS side effects, such as con-
fusion or seizures.
Famciclovir and Valacyclovir
These 2 new drugs have the same mechanism of ac-
tion as acyclovir but have the added punch of increased
drug levels after oral absorption. One study (Tyring,
1995) compared famciclovir with placebo in the treat-
ment of adults with herpes zoster and found that it re-
duced the time to lesion healing, viral shedding, and,
more importantly, the duration of post-herpetic neural-
gia by almost 2 months!
These drugs are currently indicated only for herpes
zoster and recurrent genital herpes in immunocompe-
tent adults. Adverse effects are mild and include head-
ache, nausea, diarrhea, and dizziness.
Ganciclovir
("Gang of cycles")
Fig. 29-4. A GANG OF CYCLES running over
HSV, V-Z-V, CMV, EBV, and Mr. Neutrophil and Mrs.
Platelet. GANCICLovir has broader coverage of the
herpesviridae than acyclovir, but it is more toxic.
Clinical Uses
Because of its toxicity ganciclovir is only used for
CMV infections in immunocompromised hosts:
1) AIDS patients: CMV retinitis, pneumonitis,
esophagitis.
2) Bone marrow transplant patients: CMV pneu-
monitis and prophylaxis against CMV infection.
CHAPTER 29. ANTI-VIRAL MEDICATIONS
225
Figure
29-3
Adverse effects include reversible neutropenia and
thrombocytopenia. Since zidovudine (AZT) also causes
neutropenia, carefully monitor neutrophil counts in pa-
tients taking both zidovudine and ganciclovir.
Foscarnet
This pyrophosphate analogue inhibits DNA poly-
merase and reverse transcriptase. It has extended
anti-viral activity, covering the herpesviridae and HIV.
It is important to stress that this anti-viral activity
against HIV is very minimal and not adequate for treat-
ment or viral suppression.
Foscarnet is used for AIDS patients with:
1) CMV retinitis.
2) Acyclovir-resistant strains of herpesviridae.
A big side effect, especially in AIDS patients, is re-
versible nephrotoxicity. Increased seizure potential is
possible in patients with prior history of seizure, head
trauma, renal impairment or taking concomitant med-
ications that increase seizure potential.
THE HUMAN IMMUNODEFICIENCY
VIRUS (HIV)
There has been an explosive development of new anti-
retroviral medications. "Have a HAART (Highly Active
AntiRetroviral Therapy)" is the foremost theme for those
physicians caring for HIV positive patients. HAART
refers to the use of several very potent anti-HIV (A.K.A.
antiretroviral) agents in combination to suppress viral
replication and stop the spread of resistant viruses.
There are at least 14 different anti-HIV medications in
the United States:
six
nucleoside reverse transcriptase
inhibitors f zidovudine (AZT, ZDV), didanosine (ddl), zal-
citabine (ddC), stavudine (d4T), lamivudine (3TC) and
abacavirl; three non-nucleoside reverse transcriptase in-
hibitors (nevirapine, delaviridine and efavirenz); and
five protease inhibitors (saquinavir, indinavir, ritonavir,
nelfinavir and amprenavir).
Figure 29-4
Before representing each of these drugs, let's focus on
the big picture of how to use them.
1. Antiretroviral therapy should be started for most
patients with advanced HIV infection. If their CD4
count is high and their viral burden (plasma HIV RNA
level) is very low, treatment can be delayed.
2. Three or four drugs should be used because the
data shows these combinations are more effective and
prevent emergence of resistance. Choice of agents can
be tailored to avoid side effects.
The classic principle behind HAART is the use of sev-
eral different agents with varying mechanisms of an-
tiviral activity and patterns of resistance. A three drug
combination of two nucleoside reverse transcriptase in-
hibitors and a protease inhibitor is the 1st line standard
of care. Side effects or drug interactions caused by pro-
tease inhibitors decrease their appeal in some patients.
Therefore, "protease-sparing" regimens have been de-
signed. These alternative regimens consist of two nu-
cleoside analogs and a non-nucleoside analog. Tailoring
anti-HIV medications requires patience while finding
the right mix of efficacy and tolerability. Specific exam-
ples of these combinations are shown below.
Three-drug combinations:
zidovudine + lamivudine + protease inhibitor
stavudine + lamivudine + protease inhibitor
stavudine + didanosine + protease inhibitor
Protease-sparing combinations
zidovudine + didanosine + nevirapine
zidovudine + didanosine + efavirenz
3. Physicians must follow CD4 T-lymphocyte counts,
viral load assays, and the patient's clinical status to de-
termine if treatments are effective. If CD4 counts drop,
viral load increases, or opportunistic diseases develop,
therapy should be changed. If side effects develop, drugs
should likewise be changed.
There will be many more trials in the following years
with the goal of completely suppressing viral load. HIV
may become a virus we harbor in a suppressed state and
AIDS may be prevented indefinitely. A new problem is
already emerging, especially for less developed na-
CHAPTER 29. ANTI-VIRAL MEDICATIONS
226
Figure 29-5
tions-cost. Protease inhibitor com binations cost
$10,000 to $20,000 a year! (Zuger, 1996).
NUCLEOSIDE REVERSE
TRANSCRIPTASE INHIBITORS (NRTIs)
Zidovudine (ZDV or AZT)
This is the first-line anti-HIV medication. Large stud-
ies have shown that zidovudine:
1) Reduces mortality and opportunistic infec-
tions in symptomatic HIV-infected patients with CD4
T-lymphocyte counts less than 200/mm
3
(Fischl, 1987).
2) Delays progression to AIDS in HIV infected pa-
tients with CD4 T-lymphocyte counts less than 500/mm
3
(Volberding, 1990; Fischl, 1990).
The problem with zidovudine is that HIV can rapidly
develop resistance to zidovudine when it is used alone.
This is the rationale for always starting with 2 agents.
3) Reduces maternal-to-infant transmission of
HN when given to the mother orally prior to birth, in-
travenously during delivery, and then to the baby orally
I
CDC r-HELPER CELL
RED BLOOD CELL
REUTR WHIL
GRANULOCYTE

for 6 weeks. In a recent study, this regimen reduced the
transmission rate from 25% to 8% (Conner, 1994)!
Fig. 29-5. AIDS knocks out CD4 T-lymphocytes, and
AZT (ZDV) knocks out red blood cells (anemia) and neu-
trophils (neutropenia).
It also causes, other pesky adverse effects including
headache, insomnia, myalgias, nausea, and CNS dis-
turbances (confusion, seizures). If a patient develops
these problems, the dose can be decreased or another
anti-HIV drug can be used.
Didanosine (ddl), Zalcitabine (ddC),
Stavudine (d4T), and Lamivudine (3TC)
These nucleoside reverse transcriptase inhibitors are
proving effective in reducing viral RNA load, increasing
CD4 counts, and slowing progression to AIDS. When
added to zidovudine, they prevent the emergence of zi-
dovudine resistance. The combination of zidovudine and
lamivudine (3TC) has been particularly effective and is
considered the first line of therapy when combined with
a protease inhibitor or non-nucleoside reverse tran-
scriptase inhibitor. In fact, zidovudine and lamivudine
are now available in a combination product, Combivir.
Combination products will likely be a trend for future
treatment of HIV because it decreases the number of
tablets that patients are required to take (A.K.A. pill-
burden) and increases compliance with therapy.
Lamivudine (3TC)
Lamivudine is generally well tolerated. No dose-limit-
ing toxic effects have been reported. In addition to the
treatment of HIV, lamivudine plays a very important role
in the treatment of hepatitis B virus (HBV) as monother-
apy and in combination with interferon (IFN) alpha.
Lamivudine potently inhibits hepatitis B viral DNA
replication and has a very favorable side effect profile.
Didanosine (ddl)
This is a synthetic purine nucleoside analogue that is
unstable in acid conditions, such as the gastric environ-
ment. Therefore, it is formulated with a buffer or
antacids, and should be taken on an empty stomach.
Didanosine can cause pancreatitis which may be life
threatening, in which case the drug should be dis-
continued. Other risk factors for pancreatitis, such as his-
tory of pancreatitis, alcoholism, and hypertriglyceridemia,
may increase the likelihood of developing pancreatitis.
Zalcitabine (ddC)
Unlike didanosine, zalcitabine is well absorbed from
the gastrointestinal tract. Pancreatitis occurs less com-
monly than with didanosine. Severe oral ulcers have
been reported in up to 3% of zalcitabine-treated patients.
CHAPTER 29. ANTI-VIRAL MEDICATIONS
22 7
Stavudine (d4T)
Mild increases of hepatic transaminases have also
been noted during treatment with stavudine.
Abacavir
This synthetic carbocyclic NRTI is the newest agent
in this class. Hypersensitivity reactions have been the
most concerning adverse effect, reported in approxi-
mately 5% of patients receiving abacavir. A rash is ac-
companied by systemic signs and symptoms such as
fever, fatigue, nausea, vomiting, diarrhea or abdominal
pain. These symptoms occur early and usually appear
within the first 6 six weeks of treatment. Symptoms
usually resolve rapidly after discontinuation of the
drug. It is important too remember that once abacavir
has been discontinued because of a hypersensitivity re-
action, it should not be reintroduced. More severe out-
comes, including death, have been reported to occur
when abacavir was reinstituted.
Non-specific side effects: All of these agents can
cause rash, fatigue, headaches, nausea, vomiting, diar-
rhea, abdominal pain, and insomnia. Physicians may
need to juggle these medications to find the best agent
with the least side effects.
Peripheral Neuropathy
("Remember that the D's cause peripheral
neuropathies")
The major toxic effect associated with didanosine
(ddl), zalcitabine (ddC) and stavudine (d4T) is periph-
eral neuropathy. Peripheral neuropathy usually mani-
fests with numbness or tingling of the feet, seems to be
dose related, and is generally reversible with discontin-
uation of these agents. Pre-existing neuropathy or con-
comitant use of neurotoxic medications increases the
likelihood of developing neuropathy. Therefore, you do
not want to use these agents in combination.
Non-Nucleoside Reverse Transcriptase
Inhibitors (NNRTIs)
NNRTIs bind directly and noncompetitively to the en-
zyme reverse transcriptase. They block DNA polymerase
activity by causing conformational change and disrupting
the catalytic site of the enzyme. Unlike nucleoside ana-
logues, NNRTls do not need phosphorylation to become ac-
tive, and they are not incorporated into viral DNA. When
NNRTIs are administered as a single agent or as part of an
inadequately suppressive treatment regimen, resistance
emerges rapidly. Mutations conferring resistance to one
drug in this class generally confer cross-resistance to most
other NNRTIs. Cross-resistance to nucleoside analogues
or protease inhibitors has not been observed.

Nevirapine
Nevirapine was the first NNRTI to be approved for
treating HIV infection. Nevirapine is an inducer of the
cytochrome P450 CYP3A system, including autoinduc-
tion of its own metabolism. Because of this induction po-
tential, nevirapine is avoided in combination with other
medications that are metabolized through the CYP450
system, especially antiviral medications.
Delaviridine
Delaviridine is metabolized by the cytochrome P450
system and also inhibits CYP450 activity, including its
own metabolism. This inhibition may lead to increase
plasma levels of concurrent medications metabolized
through CYP450.
Efavirenz
Efavirenz is the newest NNRTI to be approved by the
FDA. Central Nervous symptoms have been reported in
approximately 50% of patients treated with efavirenz.
These symptoms include abnormal dreams that are of-
ten dysphoric in nature as well as insomnia, dizziness,
impaired concentration, and somnolence. Symptoms
tend to diminish with continued therapy but may in-
crease with concomitant alcohol and psychoactive drug
use. You may have to stop this drug if your patient starts
dreaming about axe-wielding Elves (Efavirenz).
In vitro studies have shown that efavirenz has in-
hibitory effect on the CYP450 system. This may lead to
drug interactions similar to those with delaviridine.
Rash
Rash is a frequently reported adverse effect associ-
ated with nevirapine, delaviridine and efavirenz occur-
ring in 17%, 18% and 27% of patients in phase 111111
trials respectively (Zalalem, 1999). The rashes were
usually mild to moderate and usually occurred within
the first 4 weeks of treatment. The rash resolved with
discontinuation of the drug. Severe rashes including ul-
cerations and Stevens-Johnson syndrome (SJS) have
also been reported.
PROTEASE INHIBITORS (PIs)
HIV protease is required for production of infectious HIV
particles. Protease inhibitors inhibit this vital enzyme.
There are currently 5 protease inhibitors: saquinavir,
ritonavir, indinavir, nelfinavir and amprenavir.
Triple therapy with ZDV, ddC, and the protease in-
hibitor saquinavir, resulted in greater and longer lasting
elevations of CD4-T cell counts and greater reductions in
viral levels than 2-drug therapy. Side effects were simi-
lar for 3 and 2 drug regimens (Collier 1996).
CHAPTER 29. ANTI-VIRAL MEDICATIONS
22 8
All the PIs have navir at the end of their names. Think
of the PIs causing a viral nadir or No virus.
Saquinavir
Saquinavir was the first PI to be approved by the
FDA. The biggest problem with saquinavir has been
that a minimal amount of the drug gets absorbed when
taken orally.
Fortovase is a soft gel formulation of saquinavir with
enhanced bioavailability that has replaced hard gel
saquinavir, Invirase. Fortovase should be taken with a
meal to increase oral absorption. The main side effects
are as you might have guessed, gastrointestinal. These
effects include diarrhea, nausea, abdominal discomfort
and pain, dyspepsia and vomiting.
Indinavir
Indinavir is the opposite of saquinavir because it is
rapidly absorbed in a fasting state. If indinavir is
given with a high-fat/high-protein meal, absorption is
substantially reduced. Consumption of a light meal
(e.g. dry toast and coffee) has minimal effect on ab-
sorption. The main side effects again are gastro-
intestinal including abdominal pain, nausea, vomiting
and diarrhea. Nephrolithiasis or "kidney stones" occa-
sionally occur with renal insufficiency or acute renal
failure. Therefore caution is needed in patients with
compromised kidney function. All patients receiving
indinavir should drink at least 1.5L (48 oz.) of water
daily to ensure adequate hydration and prevent devel-
opment of kidney stones.
Ritonavir
Ritonavir is the most poorly tolerated of the four cur-
rently available PIs. The most corn only reported side
effects are gastrointestinal, including nausea, vomiting,
diarrhea and abdominal pain.
Nelfinavir
The most frequently reported side effect associated
with nelfinavir is diarrhea, noted in up to 32% of pa-
tients. Nelfinavir-associated diarrhea is generally mild
to moderate. Other reported side effects include nausea,
vomiting, abdominal pain, and rash.
Amprenavir
Amprenavir is the latest PI to be approved by the
FDA. The most frequently reported side effects are gas-
trointestinal (nausea, vomiting, diarrhea and abdomi-
nal pain); most are graded as mild to moderate. Other
reported side effects include rash, parasthesias, and de-
pressive or mood disorders.
Interleukin-2 infusion
Interleukin-2 is a cytokine released by T-lympho-
cytes that regulates the proliferation of CD4 (helper-T)
T-lymphocytes. In a recent clinical trial HIV infected
patients with CD4 counts greater that 200 cells/cc were
treated with infusions of interleukin-2. The treatment
resulted in a dramatic rise in CD4 counts from a mean
of 400 cells/cc to 900 cells/cc! There was no increase or
decrease in the viral RNA load associated with this ele-
vation of CD4 counts to a normal level. Whether this
will translate into an improved clinical outcome re-
mains to be determined (Kovacs, 1996.)
Post-Exposure (i.e., Needle Stick)
HIV Prophylaxis
After a needle-stick or other percutaneous exposure
with HIV-infected blood the risk of seroconversion is
0.3%. This risk goes up if the injury is deep, the needle
was in the patient's vein or artery, the needle had visible
blood on it, or the patient died within 60 days of the stick
(suggesting late-stage AIDS with high levels of viremia).
Treatment after an exposure with zidovudine (ZDV),
has been shown in a case-control study to reduce the risk
of seroconversion by 79%. ZDV + lamivudine (3TC) is
more active against ZDV resistant strains of HIV and the
protease inhibitors further increase HIV killing. So the
public health service has now recommended that exposed
health workers at highest risk receive triple therapy with
ZDV, 3TC, and indinavir for 4 weeks. Lower risk expo-
sures should receive ZDV and 3TC (MMWR, 1999).
MISCELLANEOUS
ANTI-VIRAL AGENTS
Amantadine
("A Man to Dine")
Amantadine has the narrowest spectrum, only in-
hibiting Influenza A, NOT B; the "A" is for "Amanta-
dine." It is thought to do this by inhibiting viral genome
uncoating in the host cell. It has minimal side effects.
Fig. 29-6. Amantadine. You have a hot dinner date
with this stud of A MAN. You meet him TO DINE at a
fancy restaurant in town; he sits at the table and takes
off his coat (uncoats). To your intense chagrin, he then
begins to blow his nose loudly and drip strings of snot
on his plate, explaining that he has a terrible flu.
If given early during an influenza A infection, aman-
tadine will decrease the duration of flu symptoms. It
also helps prevent influenza A if given prophylactically.
For example, it can be given to nursing home residents
if there is an outbreak of influenza A.
CHAPTER 29. ANTI-VIRAL MEDICATIONS
22 9
Figure 29-6
Rimantadine appears to be as effective as amanta-
dine for the prevention of influenza A. It has less CNS
side effects (anxiety and confusion) and does not require
dose adjustments in renal failure, making it a safer
agent for the elderly.
Neuraminidase Inhibitors
More recently a new class of agents, neuraminidase
inhibitors, with clinical activity against both influenza
A and B types have been introduced. These agents tar-
get neuraminidase (see Page 173), which is responsible
for cleaving the bonds between emerging virus and the
cell and therefore freeing the virus to penetrate respi-
ratory secretions and replicate. Resistance to neu-
raminidase inhibitors appears to be slow developing.
These agents are indicated for the treatment of uncom-
plicated acute illness due to influenza and will decrease
flu-symptoms by 1 to 2 days.
Oseltamivir
Oseltamivir is available as an oral tablet, which must
be started within 2 days of onset of influenza symptoms.
Oseltamivir is the only neuraminidase inhibitor used
for flu prophylaxis as well. This agent is relatively well
tolerated with dizziness, headache, fatigue, insomnia
and vertigo being the most frequent side effects occur-
ring in less than 2% of patients.
Zanamavir
Zanamavir is available as an intranasal spray and
oral inhaler and should be initiated within 2 days of on-
set of influenza symptoms. Of course, because this drug
is delivered through the upper respiratory tract, it is not
recommended in patients with severe chronic obstruc-
tive pulmonary disease (COPD), or asthma. Nose bleed
is the most characteristic side effect occurring with the
nasal spray in up to 4% of patients.

Ribavirin
Ribavirin has a wide spectrum of activity against
many DNA and RNA viruses. However it is teratogenic
in small mammals. Due to concerns about safety, it is
only used for severe respiratory syncytial virus (RSV) in-
fections in infants, for the rare case of Lassa fever (a se-
vere influenza-like illness in Africa caused by the Lassa
fever virus, in the family Arenaviridae), and for the han-
tavirus pulmonary syndrome (investigational use).
Interferon
(alpha, beta, gamma)
Human interferons are cytokines that promote a cel-
lular anti-viral state. They have been produced in large
quantities by using recombinant DNA technology.
Many studies are looking at these agents for the treat-
ment of viral infections as well as cancers.
Interferon alpha has been used to treat hepatitis C
and B infection (chronic active or persistent hepati-
tis). Treatment results in a suppression of the virus
and clinical remission. Unfortunately, when the inter-
feron is discontinued, more than half of patients will
have a relapse (50% for hepatitis B and 75-80% for
hepatitis C)!
Ribavirin has also been used in oral form to treat
hepatitis C virus (HCV) in combination with Interferon
(IFN) and as monotherapy. While ribavirin monother-
apy is successful at decreasing elevated liver enzymes
it does not eradicate virus levels in the blood and there-
fore does not appear to be the answer. However, combi-
nation therapy has been shown to improve response
rates and to minimize drug resistance and appears to
be the most promising means of treating chronic he-
patitis C to date.
Fig. 29-7. Summary of anti-viral drugs. All of these
medications have different side effects and have to be jug-
gled around to find the combination with the least adverse
efects and greatest sustained depression of viral load.
References
Balfour HH. Antiviral Drugs. N Engl J Med 340:1255-68,
1999.
Collier AC, et al. Treatment of HIV infection with saquinavir,
zidovudine, and zalcitabine. N Engl J Med 1996;334:
1011-7.
Conne EM, et al. Reduction of maternal-infant transmission
of human immunodeficiency virus type 1 with zidovudine
treatment. N Engl J Med 331:1173-80, 1994.
CHAPTER 29. ANTI-VIRAL MEDICATIONS
23 0
Danner SA, et al. A short-term study of the safety, pharmaco-
kinetics, and efficacy of ritonavir, an inhibitor of HIV-1 pro-
tease. N Engl J Med 1995;333:1528-33.
Fischl MA, et al. AIDS Clinical Trials Group: The safety and
efficacy of zidovudine (AZT) in the treatment of subjects
with mildly symptomatic human immunodeficiency virus
type 1 (HIV) infection: a double-blind, placebo-controlled
trial. Ann Intern Med 112:727-737,1990.
Fischl MA, et al. AZT Collaborative Working Group: The
efficacy of azidothymidine (AZT) in the treatment of
patients with AIDS and AIDS-related complex: a double-
blind, placebo-controlled trial. N Engl J Med 317:185-
191,1987.
Keating MR. Antiviral Agents for Non-Human Immunodefi-
ciency Virus Infections. Symposium on Antimicrobial
Agents-Parts XV. Mayo Clin Proc 74:1266-83, 1999.
Kinchington D. Recent advances in antiviral therapy. J. Clin.
Pathol. 52:89-94, 1999.
Kovacs JA, Vogel S, Albert JM, et al. Controlled trial of inter-
leukin-2 infusions in patients infected with the human im-
munodeficiency virus. N Engl J Med 1996;335:1350-6.
Markowitz M, et al. A preliminary study of ritonavir, an in-
hibitor of HIV-1 protease, to treat HN-1 infections. N.
Engl. J. Med. 1995; 333:1534-9.
Sande MA, et al. Antiretroviral therapy for adult HIV-infected
patients; recommendations from a state-of-the-art confer-
ence. JAMA 1993;270:2583-2589.
Sanford JP, Gilbert DN, et al. Guide to Antimicrobial Therapy
1996. Antimicrobial Therapy, Inc, Dallas, Texas; 1996.
Skowron G, et al. Alternating and Intermittent Regimens of
Zidovudine and Dideoxycytidine in Patients with AIDS or
AIDS-Related Complex. Ann Intern Med 118:321-329,1993.
Treanor JJ, Hayden FG, Vrooman PS, et al. Efficacy and Safety
of the Oral Neuraminidase Inhibitor Oseltamivir in treating
Acute Influenza: a randomized controlled trial. JAMA 283:
1016-1024,2000.
Tyring S, et al. Famciclovir for the treatment of acute herpes
zoster: effects on acute disease and postherpetic neuralgia.
Ann Int Med 1995;123:89-96.
Update: Provisional Public Health Service Guidelines for the
Use of Antiretroviral Agents in HIV-Infected Adults and
Adolescents MMWR 1999; 1-47.
Volberding PA, et al. AIDS Clinical Trials Group of the Na-
tional Institute of Allergy and Infectious Diseases: Zidovu-
dine in asymptomatic human immunodeficiency virus
infection: a controlled trial in persons with fewer than 500
CD4-positive cells per cubic millimeter. N Engl J Med
322:941-9,1990.
Wenzel RP. Expanding the Treatment Options for Influenza.
JAMA 283:1057-59, 2000.
Wong DKH, et al. Effect of Alpha-Interferon Treatment in Pa-
tients with Hepatitis B e Antigen-Positive Chronic Hepati-
tis B: A Meta-Analysis. Ann Int Med 119:312-323;1993.
Zelalem T, Wright AJ. Antiretrovirals. Symposium on An
timicrobial Agents. Mayo Clin Proc 74:1284-1301, 1999.
Zuger A. Treating HIV infection: New developments. Journal
Watch 1996; 16:141-2.
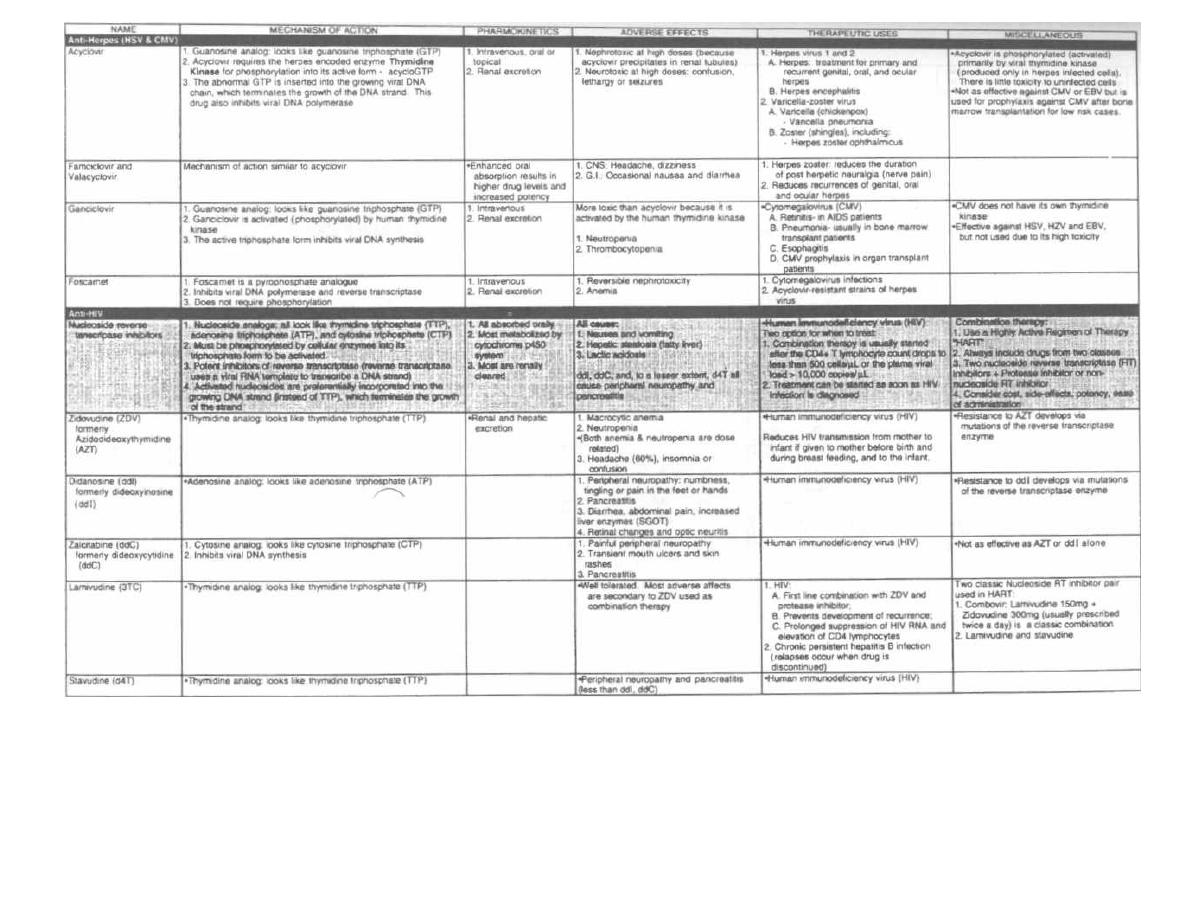
Figure 29-7
ANTI-VIRAL DRUGS
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple pMedMaster
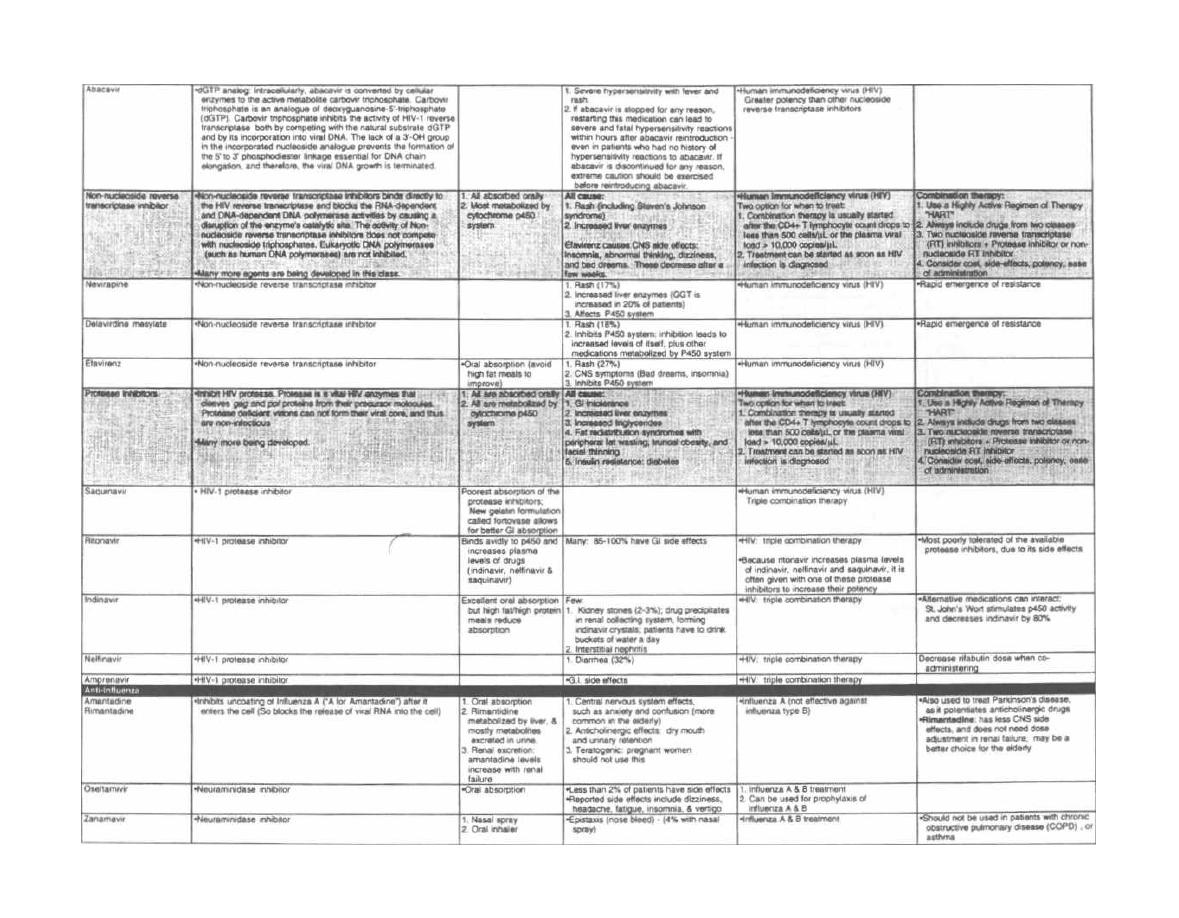
Figure 29-7 (Continued)
M. Gladwin and
a
Trattler, Clinical Microbiology Made Ridiculously Simp/e©MAedMaster

Figure 29-7 (Continued)
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple OMedMaster

Protozoa are free-living, single celled, eucaryotic cells
with a cytoplasmic membrane and cellular organelles,
i ncluding 1 or 2 nuclei, mitochondria, food vacuoles, and
endoplasmic reticulum. They come in many sizes, from
5 micrometers to 2 millimeters. They have an outer
l ayer of cytoplasm (ectoplasm) and an inner layer (en-
doplasm), which appear different from each other under
the microscope.
The protozoa ingest solid pieces of food through a
small mouth called the cytostome. For example, amoe-
bas ( Entamoeba histolitica) can ingest human red blood
cells into their cytoplasm. The protozoa reproduce asex-
ually, undergoing DNA replication followed by division
i nto 2 cells. They also reproduce sexually by the fusion
of 2 cells, followed by the exchange of DNA and separa-
tion into 2 cells again.
When exposed to new environments (such as temper-
ature changes, transit down the intestinal tract, or
chemical agents), the protozoa can secrete a protective
coat and shrink into a round armored form, called the
cyst. It is this cyst form that is infective when ingested
by humans. Following ingestion it converts back into
the motile form, called the trophozoite.
THE INTESTINAL PROTOZOA
There are 5 intestinal protozoa that cause diarrhea.
Entamoeba histolytica causes a bloody diarrhea, and Gi-
ardia lamblia and Cyclospora cayetanensis cause a
non-bloody diarrhea. Both occur in normal individuals.
Cryptosporidium and Isospora belli cause severe diar-
rhea in individuals with defective immune systems
(such as patients with AIDS).
Entamoeba histolytica
This organism is the classic amoeba we have all
heard about. It moves by extending creeping projec-
tions of cytoplasm, called pseudopodia (false feet). The
pseudopodia pull it along or surround food particles.
About 10% of the world population and 1-5% of the U.
S. population are infected with Entamoeba histolytica.
Most of these infections are asymptomatic, as the amoe-
bas live in peace inside their host carriers. These carri-
ers pass the infective form, the cyst, to other individuals
by way of the fecal-oral route. It is noteworthy that ho-
mosexual men commonly are asymptomatic carriers.
Fig. 30-1.
The motile feeding form of the amoeba is
the trophozoite, which cruises along the intestinal wall
CHAPTER 30. PROTOZOA
PART 4. PARASITES
CHAPTER 30. PROTOZOA
234
eating bacteria, other protozoa, and even human in-
testinal and red blood cells. This trophozoite can convert
to a precyst form, with two nuclei, that matures into a
tetranucleated cyst as it travels down and out the colon.
The precyst contains aggregates of ribosomes, called
chromotoid bodies, as well as food vacuoles that are
extruded as the cell shrinks to the mature cyst; it is the
mature cyst that is eaten, infecting others.
Sometimes (10% of infected individuals) the tropho-
zoites invade the intestinal mucosa causing erosions.
This results in abdominal pain, a couple of loose stools
a day, and flecks of blood and mucus in the stool. The in-
fection may become severe, with bloody, voluminous di-
arrhea.
The trophozoites may penetrate the portal blood cir-
culation, forming abscesses in the liver, followed by
spread through the diaphragm into the lung. Here the
trophozoite infection causes pulmonary abscesses and
often death (worldwide: 100,000 deaths annually).
The stool is examined for the presence of cysts or
trophozoites. Trophozoites with red blood cells in the
cytoplasm suggest active disease, while cysts or tropho-
zoites without internalized red cells suggest asympto-
matic carriage. CAT scan or ultrasound imaging of the
liver will reveal abscesses if present.
Prevention rests on good sanitation: proper disposal
of sewage and purification (boiling) of water.
Fig. 30-2.
The Metro (metronidazole) runs over
Entamoeba histolytica, Giardia lamblia, Trichomonas
vaginalis, and the anaerobic cocci and bacilli including
Bacteroides fragilis, Clostridium difficile and Gard-
nerella
vaginalis.
This drug is also called Flagyl (its
trade name) because it kills the flagellated bugs, Giar-
dia and Trichomonas.
Adverse effects of metronidazole
There is no drinking allowed on the train because it
travels rapidly and jarringly, causing stomach upset to
passengers that consume alcohol (Antabuse-disulfiram
effect). If you eat the train, as King Kong once attempted,
you would end up with a metallic taste in your mouth.
Giardia lamblia
Fig. 30-1.
Giardia lamblia exists in 2 forms: as a cyst
and as a mature, motile trophozoite that looks like a
kite.
Figure 30-1
It is estimated that 5% of U.S. adults harbor this or-
ganism, mostly asymptomatically. Outbreaks occur
when sewage contaminates drinking water. The organ-
ism is also harbored by many rodents and beavers;
campers frequently develop
Giardia lamblia
infection
after drinking from "clear" mountain streams.
After ingestion of the cyst,
Giardia lamblia
converts
to the trophozoite form and cruises down and adheres
to the small intestinal wall. The organism coats the
small intestine, interfering with intestinal fat absorp-
tion. The stools are therefore packed with fat, which has
a horrific odor! The patient will have a greasy, frothy
diarrhea, along with abdominal gassy distension and
cramps. Since
Giardia
do NOT invade the intestinal
wall, there is NO blood in the stool!!!
For diagnosis and control of
Giardia:
1) Examination of stool for cysts or trophozoites.
2) Commercial immunoassay kit to detect
Giardia
lamblia
antigens in aqueous extracts of stool specimens.
3) Sanitation measures.
Treat these patients with metronidazole (see Fig.
30-2).
Cryptosporidium
It is now apparent that this critter is everywhere!
Animals and humans are equally infected and about
CHAPTER 30. PROTOZOA
235
25% of Americans show serologic evidence of previous
infection. It can cause outbreaks of diarrhea from
contaminated municipal water sources and in infants
in day care centers. Sporadic cases can occur in
travelers.
Cryptosporidiam
is ingested as a round oocyst that
contains 4 motile sporozoites. Its life cycle occurs
within the intestinal epithelial cells, and it causes di-
arrhea and abdominal pain. These symptoms are self-
li miting in immunocompetent individuals. However,
in immunocompromised patients (AIDS patients, can-
cer patients, or organ transplant recipients who are
receiving immunosuppressive therapy), this organism
causes a severe, protracted diarrhea that is life-
threatening. These patients may have 3-17 liters of
stool per day.
Currently, there is no effective therapy. A new
macrolide drug, azithromycin (see Chapter 17), is be-
ing studied.
Isospora and Microsporidia
These organisms cause a severe diarrhea in immuno-
compromised individuals. They are transmitted via the
fecal-oral
route. Fortunately, the combination of
trimethoprim with sulfamethoxazole (see Chapter
19) is effective against
Isospora,
while albendazole
(see Chapter 31) can treat
Microsporidia.

Figure 30-2
THE SEXUALLY TRANSMITTED
PROTOZOAN
Trichomonas vaginalis
Fig. 30-3.
Trichomonas vaginalis
is transmitted sex-
ually and hangs out in the female vagina and male ure-
thra. The trophozoite of
Trichomonas vaginalis
is a
flagellated protozoon (as is
Giardia lamblia).
A female patient with this infection may complain of
itching (pruritus), burning on urination, and copious
vaginal secretions. On speculum examination you will
CHAPTER 30. PROTOZOA
236
find a thin, watery, frothy, malodorous discharge in the
vaginal vault. Males are usually asymptomatic.
Diagnosis of
Trichomonas:
1) Microscopic examination of vaginal discharge on a
wet mount preparation will reveal this highly motile
parasite.
2) Examination of urine may also reveal
Tri-
chomonas vaginalis.
Treat your patient with metronidazole (see Fig. 30-
2). Provide enough for sexual partners. Even though
males are usually asymptomatic, they must be treated

J
Figure 30-3
or the female partner will be reinfected (since this or-
ganism is not invasive, no immunity is acquired).
THE FREE-L WING MENINGITIS-
CAUSING AMOEBAS
Both
Naegleria fowleri
and
Acanthamoeba
are free-
living amoeba that live in fresh water and moist soils.
Infection often occurs during the summer months when
people swim in freshwater lakes and swimming pools
that harbor these organisms. Although large numbers
of persons are exposed, actual infection rarely occurs.
When it does, the organisms penetrate the nasal mu-
cosa, through the cribriform plate, into the brain and
spinal fluid. Both amoeba can cause an infection of the
meninges and brain (meningoencephalitis).
Naegleria
fowleri
will cause a sudden deadly infection in immuno-
competent persons, while
Acanthamoeba
will cause a
slow granulomatous infection, usually in immunocom-
promised persons.
Naegleria fowleri
Fig. 30-4.
Naegleria
fowleri is known for FOWL
PLAY, since 95% of patients will die within 1 week. In-
fected persons will present with a fever, headache, stiff
neck, nausea, and vomiting, which is very similar to a
bacterial meningitis. If asked, they will give a history of
swimming a week earlier. Examination of cerebrospinal
fluid (CSF) reveals a high neutrophil count, low glucose,
and high protein, exactly like a bacterial meningitis!!!
The Gram stain and culture will reveal NO bacteria,
CHAPTER 30. PROTOZOA
237
and microscopic examination may show the motile
amoeba.
Two patients who survived were treated with in-
trathecal amphotericin B, an antifungal agent.
Acanthamoeba
Acanthamoeba
is responsible for a chronic, granulo-
matous, brain infection in immunocompromised pa-
tients, such as those with AIDS. Over a period of weeks,
they will develop headache, fever, seizures, and focal
neurologic signs. Examination of the CSF and brain tis-
sue will reveal
Acanthamoeba
in both the cyst stage and
trophozoite stage. Treatment is difficult and involves
multiple antifungal drugs with pentamidine.
This organism may also infect the cornea (in im-
munocompetent persons), often when contact lenses are
not properly cleaned. This corneal infection (keratitis)
can lead to blindness. Treatment is with antimicrobial
eye drops.
Fig. 30-5. Comparison of Naegleria and Acan-
thamoeba infection.
THE MAJOR PROTOZOAN
INFECTIONS IN AIDS PATIENTS
The ineffective immune system in AIDS patients sets
them up for certain infections that seldom affect the im-
munocompetent host. We have already discussed 2 par-
asites that can establish a severe, chronic diarrhea in
AIDS patients:
Cryptosporidium
and
Isospora. Two

Figure 30-4
Figure 30-5
THE AMOEBAS
other parasites found more commonly in AIDS patients
than in the general population are Toxoplasma gondii
and Pneumocystis carinii. These protozoa are harbored
by most persons without problems. In AIDS, when the
T-helper cell count drops below 200, these bugs flourish
and cause disease.
Toxoplasma gondii
Many animals are infected with Toxoplasma, and hu-
mans are infected by the ingestion of cysts in under-
cooked meats (raw pork) or food contaminated with
CHAPTER 30. PROTOZOA
238
household cat feces. Kitty litter boxes are the most com-
mon source of exposure for humans, as up to 80% of cats
are infected in the United States. Toxoplasma gondii
undergoes sexual division in the cat and is excreted in
the feces as the infectious cyst.
The protozoan causes disease by reactivation of a la-
tent infection in an immunocompromised person or as a
primary infection in a pregnant woman (leading to
transplacental infection of the fetus).
1) Immunocompromised patients with AIDS or those
who are taking immunosuppressive drugs (for cancer or
Naegleria
Acanthamoeba
A
CUTE meningoencephalitis in normal
hosts
C
HRONIC meningoencephalitis in
immunocompromised hosts
M
ature amoeba only in brain tissue
(NO cysts)
C
ysts and mature amoeba found in
brain tissue
N
o corneal infection
C
orneal infection: diagnose by corneal
scraping

organ transplantation) are susceptible to growth of the
latent
Toxoplasma gondii.
The infection can present in
many ways-with fever; lymph node, liver, and spleen
enlargement; pneumonia; or frequently with infection of
the meninges or brain. In fact,
Toxoplasma
encephalitis
is the most common central nervous system infection
in AIDS patients. The brain infection can involve a
growing mass, much like a tumor, with symptoms of
headache and focal neurologic signs (seizures, gait in-
stability, weakness, or sensory losses). Infection of the
retina, chorioretinitis, is also common, resulting in vi-
sual loss. Examination of the retina reveals yellow-
white, fluffy (like cotton) patches that stand out from
the surrounding red retina.
2) Toxoplasma
is one of the transplacentally ac-
quired TORCHES organisms that can cross the blood-
placenta barrier (see Fig. 26-2.). Transplacental fe-
tal infection can occur if a pregnant woman who has
never been previously exposed to
Toxoplasma gondii
is infected. Congenital toxoplasmosis does not occur
in pregnant women who have serologic evidence of
previous exposure, most likely because of a protec-
tive immune response. Pregnant women should
avoid cats!!
Like rubella (see Chapter 28), congenital toxoplasmo-
sis can cause many problems, including chorioretinitis,
blindness, seizures, mental retardation, microcephaly,
encephalitis, and other defects. If the infection is ac-
quired early during gestation, the disease is severe, of-
ten resulting in stillbirth. Interestingly, infants that
appear normal can develop disease later in life. Clinical
reactivation results most commonly in retinal inflam-
mation (chorioretinitis, which can result in blindness)
that flares late in life (peak incidence in second or third
decade).
Note that immunocompetent adults (such as the
pregnant women described above) who are infected with
Toxoplasma gondii
often develop generalized lymph
node enlargement.
BIG PICTURE: In AIDS patients and fetuses
Toxoplasma gondii is
TOXIC
to the
BRAIN
and
EYES!!!
Diagnosis of toxoplasmosis can be made by:
1) CAT scan of brain will show a contrast-enhanc-
ing mass.
2) Examination of the retina of the eye will reveal
retinal inflammation.
3) Serology: Elevated immunoglobulin titers suggest
that the patient has at some time been exposed to this
organism.
Sulfadiazine plus pyrimethamine can be used to
treat patients with acute toxoplasmosis.
CHAPTER 30. PROTOZOA
23 9
Pneumocystis carinii
Pneumocystis carinii
is a flying-saucer appearing
FUNGUS that has previously been classified as a proto-
zoan but now has been shown to be more closely related
to fungi. This organism appears to invade the lungs of all
individuals at an early age and persists in a latent state.
In fact, based on IgM and IgG levels, it appears that
about 85% of children have had a mild or asymptomatic
respiratory illness with
Pneumocystis carinii
by age 4. In
persons with a functioning immune system, this organ-
ism will live comfortably within the lung without caus-
ing symptoms. However, in immunocompromised pa-
tients (AIDS patients, cancer patients, and organ trans-
plant recipients), this organism can multiply in the
lungs and cause a severe interstitial pneumonia, called
Pneumocystis carinii
pneumonia (PCP).
PCP is the most common opportunistic infection of
AIDS patients. Without prophylactic treatment there is
a 15% chance each year of infection, if the CD4+ T-
helper cell count is below 200. Clinically, this illness
presents with fever, shortness of breath, a nonproduc-
tive cough, and eventually death if not properly treated.
Chest X-ray may show diffuse bilateral interstitial in-
filtrates, or it can be normal.
The Case of the Breathless Woman Who Had
No Helpers
A 22 year-old female comes to the hospital with fever
and a feeling of chest tightness. She says she has no
medical problems but has lost weight. On physical ex-
amination you find large lymph nodes everywhere and
numerous genital warts. You note that she is tachy-
pneic, breathing 30 breaths per minute.
You look over her past record and find that she had a
child that was born with AIDS.
Her chest film shows diffuse perihilar interstitial
streaking bilaterally, sparing the outer lung margins.
You order an absolute T4-cell count and find that she
has 150 T-helper cells (Normal is greater than 1000).
Diagnosis of
Pneumocystis carinii
can be made by
silver-staining alveolar lung secretions, revealing the
flying saucer-appearing fungi. These secretions can be
obtained as follows, in order of increasing yield:
1) Induce a sputum sample by spraying saline into
the bronchioles and collecting the coughed material
(60% sensitivity).
2) Insert a fiberoptic camera (bronchoscope) deep
into the patient's bronchial tree, inject saline, and then
wash it out (bronchoalveolar lavage) (98% sensitivity).
3) Biopsy the lung by bronchoscopy (100% sensitivity).
About 80% of AIDS patients will get PCP at least
once in their lifetime unless prophylactic trimetho-
CHAPTER 30. PROTOZOA
Figure 30-6
prim/sulfamethoxazole is given when
CD4+
T-cell
counts drop below
200-250.
More than 60% of PCP in-
fections are being prevented with this prophylactic in-
tervention! Symptomatic patients can be treated with
high dose trimethoprim/sulfamethoxazole or intra-
venous pentamidine.
MALARIA
Plasmodium falciparum, Plasmodium vivax,
Plasmodium ovale,
and
Plasmodium malariae
Malaria is a febrile disease caused by
4
different pro-
tozoa:
Plasmodium falciparum, Plasmodium vivax, Plas-
modium ovale,
and
Plasmodium malariae.
They infect
about 300-500 million persons worldwide each year, re-
sulting in
20-40
million deaths. The anopheles mosquito
carries the organisms within its salivary glands and in-
jects them into humans while it feeds. The organisms
then grow in the liver and spread to the human red blood
cells, where they reproduce. The red cells fill with proto-
zoa and burst. The red cells all burst at the same time, re-
leasing the protozoa into the bloodstream and exposing
them to the immune system, which results in fever.
Fig. 30-6. The different species of
Plasmodium
burst the red cells at different time intervals.
Plus-
240
modium, vivax
and
Plasmodium ovale
burst loose
every
48
hours. The people who discovered all this
started numbering things with one (they didn't have a
zero) and so zero hours was called one,
24
hours
called two, and
48
hours called three. So, P.
vivax
and P.
ovale
produce chills and fever followed by
drenching sweats every
48
hours, which is called
tertian malaria. P.
malariae
bursts loose every 72
hours, causing a regular 3-day cycle of fevers and
chills, followed by sweats. This is called quartan
malaria. P.
falciparum,
the most common and deadly
of the
Plasmodia,
bursts red cells more irregularly,
between 36
48
hours. Thus the chills and fevers tend
to either fall within this period or be continuous, with
less pronounced chills and sweats.
Plasmodia Life Cycle
Fig. 30-7. The life cycle of the
Plasmodia
is complex,
since the protozoa divide into many different forms with
different names. This is clinically important because
the diagnosis of this disease rests on being able to iden-
tify these forms on a slide of a patient's blood.
Imagine yourself in Kenya with a patient who has in-
termittent shaking chills, fevers, and soaking sweats.
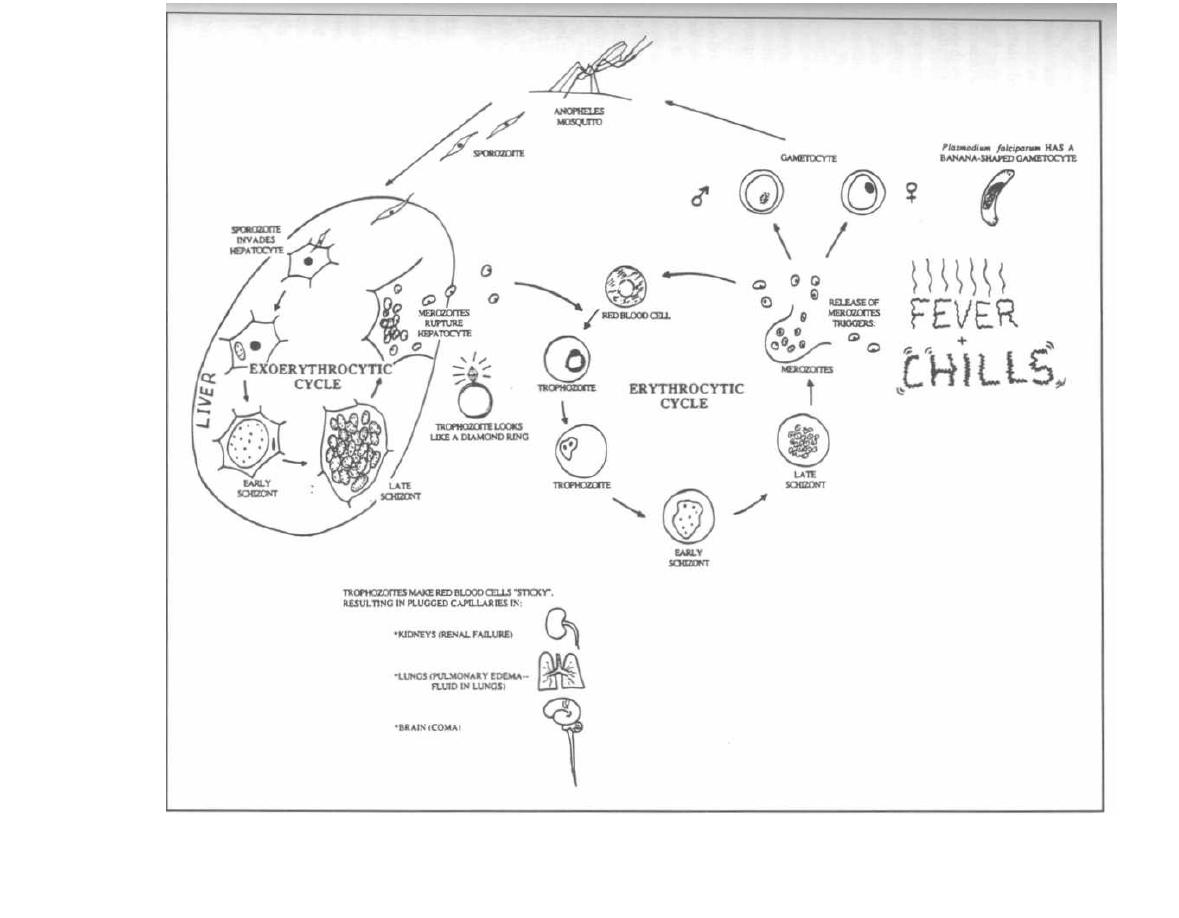
Figure 30-7

You pull out your little microscope with a reflecting mir-
ror light source, tilt it to catch some of the deep red
African sun, and focus your attention on the smear of
red cells in front of you. What are those life cycle stages
and what do they look like?
Plasmodia
undergo sexual division in the anopheles
mosquito and asexual division in the human liver and
red cells. Let's start with the human:
Thin, motile, spindle-shaped forms of the
Plasmodia,
called sporozoites, swim out of the mosquito's sucker
and into the human bloodstream. They wiggle their way
to the liver and burrow into a liver cell. This marks the
beginning of the pre-erythrocytic cycle in the liver,
so-named because this cycle occurs before the red blood
cells (erythrocytes) are invaded. The sporozoite rounds
up to form a ball within the liver cell. This ball, now
called a trophozoite, undergoes nuclear division, form-
ing thousands of new nuclei. This big mass is now a cell
with thousands of nuclei, called a schizont. A cytoplas-
mic membrane then forms around each nucleus, creat-
i ng thousands of small bodies called merozoites. The
new overloaded liver cell bursts open, releasing the
merozoites into the liver and bloodstream. Some will re-
i nfect other liver cells as the sporozoite did initially, re-
peating the same cycle discussed above, which is now
called the exo-erythrocytic cycle.
Other merozoites will enter the bloodstream and
enter red blood cells, starting the erythrocytic cycle.
This cycle is similar to the exo-erythrocytic cycle,
except that it occurs in the erythrocytes rather than
the liver cells. The merozoite rounds up to form a
trophozoite. In the red cells the trophozoite is shaped
like a ring with the nuclear material looking like the
diamond on the ring. Nuclear division then occurs with
formation of a large multinucleated schizont. Cyto-
plasm surrounds each nucleus to form new merozoites
within the late schizont. Red cell lysis occurs with
release of merozoites. The released merozoites stimu-
late an immune response, manifested as fevers, chills,
and sweats.
The merozoites can continue to invade other red cells
and then grow for another 2-3 day cycle followed by rup-
ture and release again. Some merozoites will change
into male and female gametocytes. These cells circu-
late and will be taken up by a biting anopheles mos-
quito. If they are not, they will shortly die.
Two of the species, P.
vivax
and P.
ovale,
produce dor-
mant forms in the liver (hypnozoites) which can grow
years later, causing relapsing malaria. This is why you
are asked if you have ever had malaria when you donate
blood (an effort to screen out infected blood).
In the mosquito, the gametocytes are sucked into the
stomach where the male and female gametocytes fuse.
DNA is mixed and the fused gametocytes become an
oocyst. The oocyst divides into many spindle-shaped
wiggling sporozoites, which disseminate within the
mosquito. They may find their way into the mosquito
salivary gland and will be injected into the human for
asexual reproduction.
The Disease Malaria
Malaria is well known for causing periodic episodes of
severe chills and high fevers along with profuse sweat-
ing at 48-72 hour intervals. These episodes commonly
last about 6 hours and are associated with the rupture
of red blood cells.
You can imagine that all these cycles of red blood cell
lysis must take their toll! In fact, P.
falciparum,
the
most aggressive species, will invade up to 30% of ery-
throcytes, which results in anemia and sticky red blood
cells. These sticky cells plug up post-capillary venules in
organs such as the kidney, lung, and even brain, result-
ing in hemorrhages and blocked blood delivery to those
tissues. Renal failure, lung edema, and coma may en-
sue, leading to death. Most deaths occur in children less
than 5 years old in sub-Saharan Africa. These children
often develop cerebral malaria characterized by
seizures and impaired consciousness, leading to coma.
Even with treatment, 20% of children with cerebral
malaria will die.
Infected individuals also get hepatomegaly and
splenomegaly. The spleen and liver enlarge as the fixed
phagocytic cells (of the reticuloendothelial system) pick
up large amounts of debris from the destroyed red blood
cells. The enlarged spleen occasionally ruptures.
Many African-American and African blacks are resis-
tant to P.
vivax
and
P. falciparum
infection. The resis-
tance to P.
vivax
is based on the absence of red cell
membrane antigens Duffy a and b that the P.
vivax
uses for binding. The sickle cell anemia trait (hemoglo-
bin AS) appears to help protect the red cells from P.
Figure 30-8
MALARIA
CHAPTER 30. PROTOZOA
242

I
falciparum
invasion. Endemic infection with malaria in
the African continent is thought to have led to a Dar-
winian selection process, resulting in high levels of
sickle trait and absence of Duffy a and b in many
African and African-American blacks.
Fig. 30-8.
Comparison of the
Plasmodia
species.
Diagnosis
1) Examination of thin and thick smears
(1000x)
of
blood, under oil-immersion magnification, reveals the
trophozoites and schizonts within the erythrocytes.
Sometimes the gametocytes can be visualized.
2) Fluorescently labeled antibodies may be used to
identify the responsible species.
Control of Malaria
1) Prevent mosquito bite:
a) Eliminate vector with pesticides (pyrethins) at
dusk in living and sleeping areas.
b) Use insect repellants (containing DEFT) and
mosquito nets, and wear long-sleeved shirts and
long pants.
2) Chemical Prophylaxis for travelers: When traveling
to an area without chloroquine resistance, chloroquine
is used. If in a chloroquine resistant area, mefloquine or
doxycycline may be used for prophylaxis.
It is wise to carry a pyrimethamine/sulfadoxine (fan-
sidar) "starter pack" to take in case of breakthrough in-
fection when far away from medical care. This is
especially true with doxycycline.
Fig. 30-9.
Chloroquine-resistant
P. falciparum
ar-
eas. Malaria is a disease of the tropics, cutting a swath
across the equator. Chloroquine-resistant
Plasmodium
falciparum
areas include most of Africa, Central Amer-
ica south of the Panama Canal, South America, India,
and South East Asia (see map). Chloroquine-sensi-
tive areas include North Africa, Central America North
of the Panama Canal, Haiti, and the Middle East.
Treatment of Malaria
To treat malaria you must understand two concepts:
1) the geographic pattern of susceptibility of P.
falci-
parum
to antimalarial drugs ( Fig.
30-9.)
and 2) the type
of
Plasmodium
species causing the infection.
1) P. malariae, P. vivax,
and P.
ovale
are all suscepti-
ble to chloroquine! Pushovers! But don't forget that P.
vivax and P.
ovale
have exo-erythrocytic cycles in the
liver and will be protected there from chloroquine. The
CHAPTER 30. PROTOZOA
243
Figure 30-9
acute infection will subside, but relapses will occur.
Treatment with
Primaquine
will kill the liver holdouts.
Primaquine is the Prime drug to kill
P. vivax
and P.
orale
in the liver!
2) Plasmodium falciparum:
This guy is nasty, caus-
ing the most hemolysis, organ damage, and death.
a) Chloroquine-sensitive areas: Huh, I wonder
which drug to use?? Chloroquine alone is enough
as P.
falciparum
does not have an exo-erythrocytic
cycle.
b) Chloroquine-resistant areas: Treatment options
include quinine (quinidine, the antiarrhythmic
drug, is more expensive but readily available in the
United States and just as effective), artemether
(see below), pyrimethamine/sulfadoxine, or
mefloquine. Severe infection (cerebral malaria) is
treated with IV or IM quinine, quinidine, or
artemether.
Newsflash!!! Artemether is new therapy for se-
vere falciparum malaria in children and adults!
Chloroquine-resistant P.
falciparum
causes severe
malaria in Africa, killing about 500,000 children a year
(1-2 million world-wide). Quinine is the preferred ther-
apy in these areas because it can be injected intramus-
culary (IM). A new drug named artemether (or its
brother artesunate) is derived from a traditional Chi-
nese malaria remedy (qinghaosu or wormwood!).
Artemether is effective against chloroquine-resistant P.
falciparum
and has proven to work as well as quinine in
the treatment of severe malaria. Unfortunately, even
with therapy, 20% of children with cerebral malaria still
die. (Hoffman, 1996; Van Hensroek, 1996; Hien, 1996;
White, 1996).
There are a number of common features of these
drugs (also see Fig.
30-13).
1) All of the anti-malarial drugs can be taken orally.
2) All of the anti-malarial drugs cause GI upset as a
primary adverse effect.
3) Chloroquine, primaquine, and quinine all cause
hemolysis in patients with glucose-6-phosphate dehy-
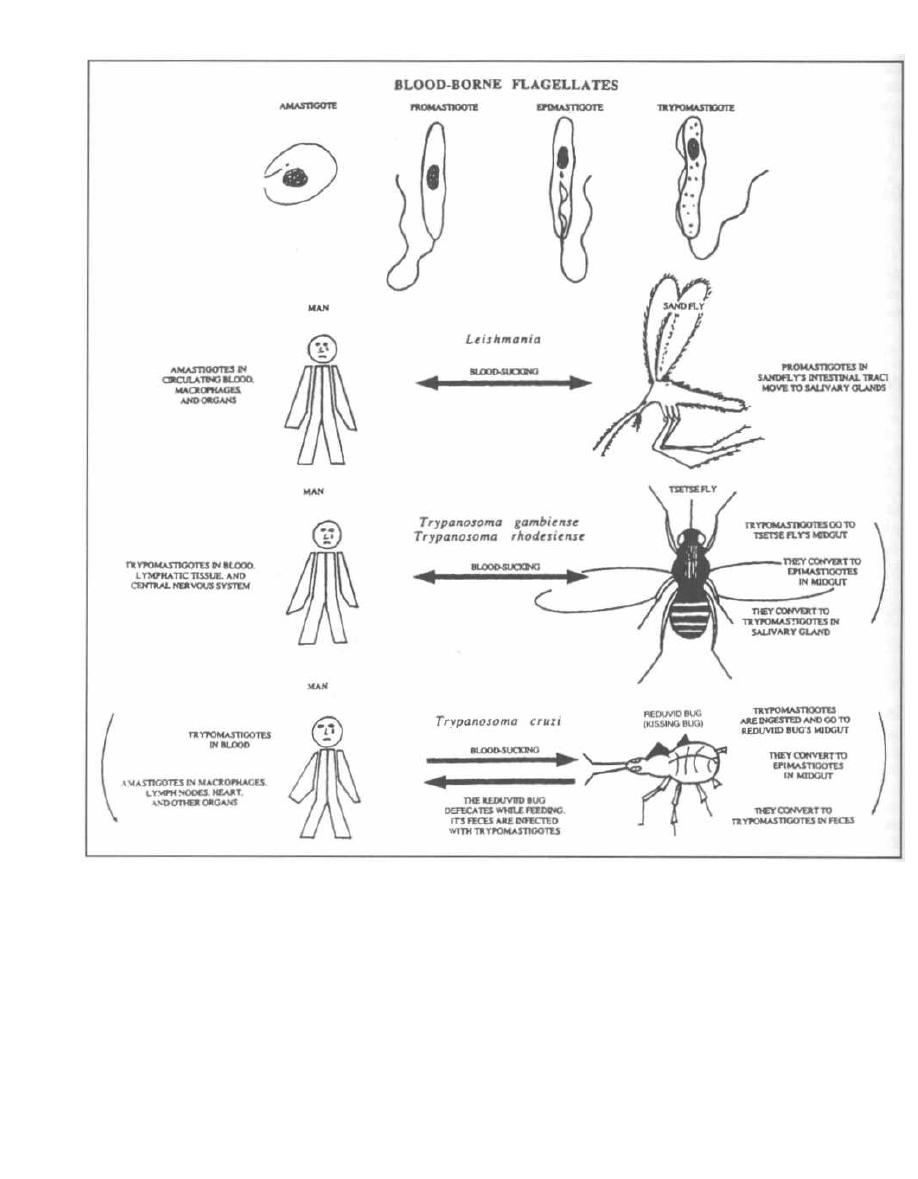
Figure 30-10
drogenase deficiency (G-6-P-D deficiency is present
in some Africans, Mediterraneans, and Southeast
Asians).
4) Chloroquine, quinine, quinidine, and sulfadoxine/
pyrimethamine are safe in all trimesters of
pregnancy. Not enough data is available about the
others.
CHAPTER 30. PROTOZOA
24 4
BABESIOSIS
( Babesia microti, Babesia divergens )
Babesiosis is an infection very much like malaria. It is
transmitted by the bite of a blood sucker (tick in this case)
and it invades and can be seen inside, red blood cells. It
also causes fever and hemolysis (anemia), as in malaria.

It is different in that:
1) There are more than 100 species of Babesia, mostly
causing disease in cattle and other domestic or wild
animals.
2) Babesia are spread by tick bites, not mosquito bites.
3) They do not affect liver cells (so there is no exoery-
throcytic phase).
In the northeastern coastal United States (e.g. Nan-
tucket Island) Babesia Microti is spread by the bite of
the same tick that spreads lyme disease, Ixodes scapu-
laris. After biting the white-footed mouse, the reservoir
for B. microti, the tick will leap to the next carefree
golfer who walks into the rough.
Like Plasmodium, Babesia sporozoites slither out of
tick salivary glands into the blood of the hapless golfer.
The sporozoites invade erythrocytes and differentiate
into pear or ring-shaped trophozoites. Trophozoites
asexually bud and divide into 4 merozoites that stick to-
gether, forming a cross or x-shaped tetrad ("Maltese
cross"). Red cell infection results in only mild hemolysis,
so infection is usually asymptomatic and sub-clinical.
Asplenic patients are unable to clear the organisms as
well and may have severe infection similar to falci-
parum malaria. (Gelfand, 1995; Persing, 1995). Treat
infected patients with quinine and clindamycin.
Fig. 30-9A. Babesia sporozoites in the "hood".
Giemsa or wright-stained thin and thick blood smears
reveal ring-shaped trophozoites that look like Plasmod-
ium and the distinctive cross or x-shaped tetrad of
merozoites (Maltese cross). Transmitted by tick vector.
THE BLOOD-BORNE FLAGELLATES
(Leishmania and Trypanosoma)
Fig. 30-10. Leishmania and Trypanosoma are trans-
mitted by the bite ofa blood-sucking insect. Although they
CHAPTER 30. PROTOZOA
245
cause different diseases, they pass through similar mor-
phologic states in the human and insect host. They can ex-
ist as rounded cells without flagella, called amastigotes,
or as flagellated motile forms called promastigotes, epi-
mastigotes, and trypomastigotes. These are named
according to the insertion site oftheir single flagellum. All
ofthese organisms cause an initial skin ulcer at the site of
the insect bite, followed by systemic invasion.
These parasites can also be transmitted via blood
product transfusion.
Leishmaniasis
(Leishmania tropica, Leishmania chagasi, Leishmania
major, Leishmania braziliensis, Leishmania donovani)
Leishmania is zoonotic, carried by rodents, dogs, and
foxes, and is transmitted to humans by the bite of the
sandfly. The disease leishmaniasis is found in South
and Central America, Africa, and the Middle East.
Following transmission from the sandfly, the pro-
mastigote invades phagocytic cells (macrophages) and
transforms into the nonmotile amastigote. The amastig-
ote multiplies within the phagocytic cells in the lymph
nodes, spleen, liver, and bone marrow (the reticuloen-
dothelial system) (see Fig. 30-10).
The different diseases caused by Leishmania depend
on the invasiveness of the species as well as the host's
immune response. Host immunity depends on a cell-me-
diated defense. It appears that some patients have ge-
netically deficient defenses against Leishmania and
will be afflicted with more severe disease. Leishmania-
sis presents in a spectrum of disease severity: from a
single ulcer that will heal without treatment; to widely
disseminated ulcerations of the skin and mucous mem-
branes; to the very severe infection striking deep into
the reticuloendothelial organs, the spleen and liver.
Note the similarity here to leprosy (see Chapter 14) in
which differences in host cell-mediated defenses result
in varied severity of disease.
There are 3 clinical forms of this disease:
1) Cutaneous leishmaniasis
a) Simple cutaneous lesions
b) Diffuse cutaneous lesions
2) Mucocutaneous leishmaniasis
3) Visceral leishmaniasis
Cutaneous Leishmaniasis
A sandfly injects Leishmania into the skin, where
they migrate to reticuloendothelial cells (fixed phago-
cytic cells in lymph nodes). At the site of the sandfly
bite, a skin ulcer develops, called an "oriental sore." This
ulcer heals in about a year, leaving a depigmented (pale)
scar. Diagnosis is made by observing Leishmania in
stained skin-scrapings from the ulcer base.
Cell-mediated immunity is intact, so the immune sys-
tem attacks the organisms resulting in skin destruction
(ulcer formation) and clearance of infection (similar to
the situation with tuberculoid leprosy). Because of the
intact cell-mediated immunity, this organism invokes a
delayed hypersensitivity reaction. Diagnosis can be
made by injecting killed
Leishmania
intradermally
(Leishmania skin test). Just like the PPD test of tuber-
culosis, a raised indurated papule 48 hours later sup-
ports the presence of a
Leishmania
infection.
This disease is also seen in Latin America and Texas,
where it is called American cutaneous leishmaniasis.
Diffuse Cutaneous Leishmaniasis
In Venezuela and Ethiopia, a chronic form of cuta-
neous leishmaniasis occurs in patients with deficient
immune systems. A nodular skin lesion arises but does
not ulcerate. With time, numerous nodular lesions arise
diffusely across the body. There is often a concentration
of lesions near the nose. The untreated infection can last
more than 20 years.
The disease is diffuse because the host's immune
system does not respond to the invasion by
Leishma-
nia,
due to a defect in cell-mediated immunity. There-
fore, the promastigotes are able to spread and infect
the skin, causing the diffuse nodular lesions. Due to
the defect in cell-mediated immunity, the Leishmania
skin test is negative (similar to the situation in lepro-
matous leprosy).
Mucocutaneous Leishmaniasis
Initially, a dermal ulcer, similar to cutaneous leishma-
niasis, arises at the site of the sandfly bite and soon heals.
However, months to years later, ulcers in the mucous
membranes of the nose and mouth arise. If untreated, the
infection is chronic, with erosion of the nasal septum, soft
palate, and lips, over a course of 20-40 years. Death by
bacterial secondary infection may occur.
Diagnosis is made via skin scrapings.
Visceral Leishmaniasis (Kala-azar)
The sandfly transmits
Leishmania donovani
or
Leish-
mania chagasi
to an individual (most commonly young
malnourished children), who months later will complain
of abdominal discomfort and distension, low-grade
fevers, anorexia, and weight loss. This abdominal en-
largement is due to
Leishmania donovani's
invasion of
the reticuloendothelial cells (fixed phagocytic cells) of the
spleen and liver, causing hepatomegaly and massive
splenomegaly. Patients also develop a severe anemia
and the white blood-cell count can also be very low. Most
cases (over 90%) are fatal if untreated.
Diagnosis is made by liver and spleen biopsies
demonstrating these protozoa. The Leishmania skin
CHAPTER 30. PROTOZOA
246
test is negative during active disease as cell-mediated
immunity is deficient.
All forms of leishmaniasis can be treated with the
pentavalent antimonial stibogluconate.
African Sleeping Sickness
(Trypanosoma brucei rhodesiense
and
Trypanosoma
brucei gambiense)
Trypanosoma rhodesiense
and
Trypanosoma gambi-
ense
are responsible for African sleeping sickness, which
is transmitted by the blood-sucking bite of a tsetse fly.
Following this bite, the motile flagellated form of these
2 organisms, called a trypomastigote, spreads via the
person's bloodstream to the lymph nodes and central
nervous system (CNS) (see Fig. 30-10).
The first manifestation is a hard, red, painful skin ul-
cer that heals within 2 weeks. With systemic spread, the
patient then experiences fever, headache, dizziness, and
lymph node swelling. These symptoms can last a week,
and then the fever subsides for a few weeks followed by
renewed fevers. This pattern of fevers with fever-free in-
tervals can occur for months. Finally, CNS symptoms
develop, with drowsiness in the daytime (thus sleeping
sickness), behavioral changes, difficulty with walking,
slurred speech, and finally coma and death.
There are 2 forms of African sleeping sickness. West
African sleeping sickness, caused by
Trypanosoma
brucei
gambiense, is notable for slowly progressing
fevers, wasting, and late neurologic symptoms. East
African sleeping sickness, caused by
Trypanosoma
brucei rhodesiense,
is similar to the West African vari-
ety but more severe, with death occurring within weeks
to months. There is rapid progression from recurrent
fevers to early neurologic disease (drowsiness, mental
deterioration, coma, and death).
Q: Why the intermittent fevers???
A: Variable surface glycoproteins (VSG). The
trypanosomes are covered with about 10 million mole-
cules of a repeating single glycoprotein called the VSG.
The trypanosomes possess genes that can make thou-
sands of different VSGs. They will make and express, on
their surface, a new VSG in a cyclical nature. Every
time the human host develops antibodies directed
against the VSG (and fever with this immune recogni-
tion), the trypanosomes produce progeny with a new
VSG coat. Thus, there are waves of new antigens, pro-
ducing recurrent fevers and protection from our im-
mune defenses. This is similar to the antigenic variation
of the spirochete
Borrelia recurrentis,
which causes re-
lapsing fever (see Fig. 13-12).
Diagnosis consists of visualization of trypomastigotes
in peripheral blood, lymph nodes, or spinal fluid.

Figure 30-11
Patients are treated with suramin if the central ner-
vous system (CNS) is not involved (suramin does not
penetrate into the CNS). With CNS involvement, the ar-
senical melarsoprol, which is extremely toxic, is used.
Chagas' Disease
(Trypanosoma cruzi ,
the American Trypanosome)
Chagas' disease is caused by a trypanosome, but the
pathogenesis and epidemiology differ greatly from the
African trypanosomes.
This is truly a disease of the Americas, ranging from the
southern U. S. (Texas), Mexico, Central America, and
down into South America.
T. cruzi
survives in wild animal
reservoirs such as rodents, opossums, and armadillos.
The vector is the reduviid bug, also called the kissing
bug. The bug feeds on humans while they sleep and defe-
CHAPTER 30. PROTOZOA
24 7
Cates while it eats.
T. cruzi
trypo-mastigotes, which are
present in the bug's feces, tunnel into the human host.
The trypomastigote loses its undulating membrane and
flagellum and rounds up to form the amastigote, which
rapidly multiplies. Organisms invade the local skin,
macrophages, lymph nodes, and spread in the blood to dis-
tant organs (see Fig. 30-10).
Acute Chagas' Disease
At the skin site of parasite entry, a hardened, red
area develops, called a chagoma. This is followed by
systemic spread with fever, malaise, and swollen
lymph nodes. Organs that can be infected include the
heart and central nervous system (CNS). Heart in-
flammation results in tachycardia and electrocardio-
graphic changes, while the CNS involvement can

result in a severe meningoencephalitis (usually in
young patients).
This acute illness resolves in about a month and pa-
tients then enter the intermediate phase. In this
phase there are no symptoms, but there are persistently
low levels of parasite in the blood as well as antibodies
against T. cruzi. Most persons will remain in the inter-
mediate phase for life.
For reasons that are poorly understood, some per-
sons will develop chronic Chagas' disease years to
decades later.
Chronic Chagas' Disease
The organs primarily affected are the heart and some
hollow organs such as the colon and esophagus. Intra-
cellular T. cruzi amastigotes cannot usually be found,
and it is unclear why disease develops in these organs.
1) Heart: Arrhythmias are the earliest manifesta-
tion (heart block and ventricular tachycardia). Later
there is an increase in heart size and heart failure (di-
lated cardiomyopathy).
2) Megadisease of colon and esophagus: A big,
dilated, poorly functioning esophagus develops with
symptoms of difficulty and pain in swallowing, and re-
gurgitation of food. A dilated colon (megacolon) results
in constipation and abdominal pain. Patients can go
weeks without bowel movements.
Fig. 30-11. Trypanosoma cruzi (the American Try-
panosome). To remember that T. cruzi causes mega-
colon, electrical arrhythmias ,and dilatation of the
heart, and is transmitted by the feces of the kissing bug,
picture Tom Cruise (the American actor).
Diagnosis and Treatment
Acute Chagas' disease:
1) Direct examination of the blood for the motile try-
pomastigotes.
2) Xenodiagnosis: This sensitive test is conducted
as follows. Forty laboratory-grown reduviid bugs are al-
lowed to feed on the patient, and one month later the
bugs' intestinal contents are examined for the parasite.
Chronic Chagas' disease:
Classic clinical findings (cardiac and megadisease)
along with serologic evidence of past T. cruzi infection
allows for presumptive diagnosis.
Although nifurtimox and benznidazole can be used
for acute cases, there is currently no effective therapy for
the chronic manifestations of this infection. Therefore,
CHAPTER 30. PROTOZOA
248
individuals should take precautions to prevent kisses by
the kissing bug (insect repellent, bednets).
Balantidium Coli
If you do not want diarrhea, do not consume food or
water contaminated by pig feces! This advice will pre-
vent ingestion of
B.
coli cysts. These cysts mature into
ciliated trophozoites, and travel to the intestinal tract.
The trophozoites dig into the intestinal wall, where they
exist happily consuming the native bacteria. Most in-
fected individuals are asymptomatic, while some will
develop diarrhea.
B. Coli
trophozoites are notable for being the largest
parasitic protozoans found in the intestine. Diagnosis is
made by identifying the ciliated trophozoites or cysts in
stool specimens. Tetracycline is effective at treating this
infection.
Fig. 30-12.
Summary of the protozoan diseases.
Fig. 30-13. Treatment of protozoan diseases.
References
Dupont HL, Chappell CL, et al. The infectivity of
Cryp-
tosporidium paruum in healthy volunteers. N Engl J Med
1995;332:855-9.
Gelfand JA. Babesia. In: Mandell GL, Bennett JE, Dolin R,
eds. Principles and Practice of Infectious Disease. 4th ed.
New York: Churchill Livingstone, 1995:2415-2427.
Hein TT, Day NP, et al. A controlled trial of artemether or qui-
nine in Vietnamese adults with severe falciparum malaria.
N Engl J Med 1996;335:76-83.
Hoffman SL. Artemether in severe malaria-still too many
deaths. N Engl J Med 1996;335:124-5.
Kirchhoff LV. American trypanosomiasis (Chagas' disease) -a
tropical disease now in the United States. N Engl J Med
1993;329:639-644.
Krogstad DJ. Plasmodium species (malaria). In: Mandell GL,
Bennett JE, Dolin R, eds. Principles and Practice of Infec-
tious Disease. 4th ed. New York: Churchill Livingstone,
1995:2415-2427.
Persing DH, Herwaldt BL, et al. Infection with a Babesia-like
organism in northern California. N Engl J Med 1995;
332:298-303.
Rosenblatt JE. Antiparasitic Agents. Symposium on Antimi-
crobial Agents-Part VII. Mayo Clin Proc 67:276-287, 1992.
Sanford JP, Gilbert DN, et al. Guide to Antimicrobial Therapy
1996. Antimicrobial Therapy, Inc, Dallas Texas; 1996.
Ungar BL. Cryptosporidium. In: Mandell GL, Bennett JE,
Dolin R, eds. Principles and Practice of Infectious Disease.
4th ed. New York: Churchill Livingstone, 1995:2500-2510.
Van Hensbroek MB, Onyiorah E, et al. A trial of artemether
or quinine in children with cerebral malaria. N Engl J Med
1996;335:69-75.
White NJ. Current concepts: The treatment of malaria. N
Engl J Med 1996;335:800-06.
Wyler DJ. Malaria chemoprophylaxis for the traveler. N Engl
J Med 1993;329:31-37.
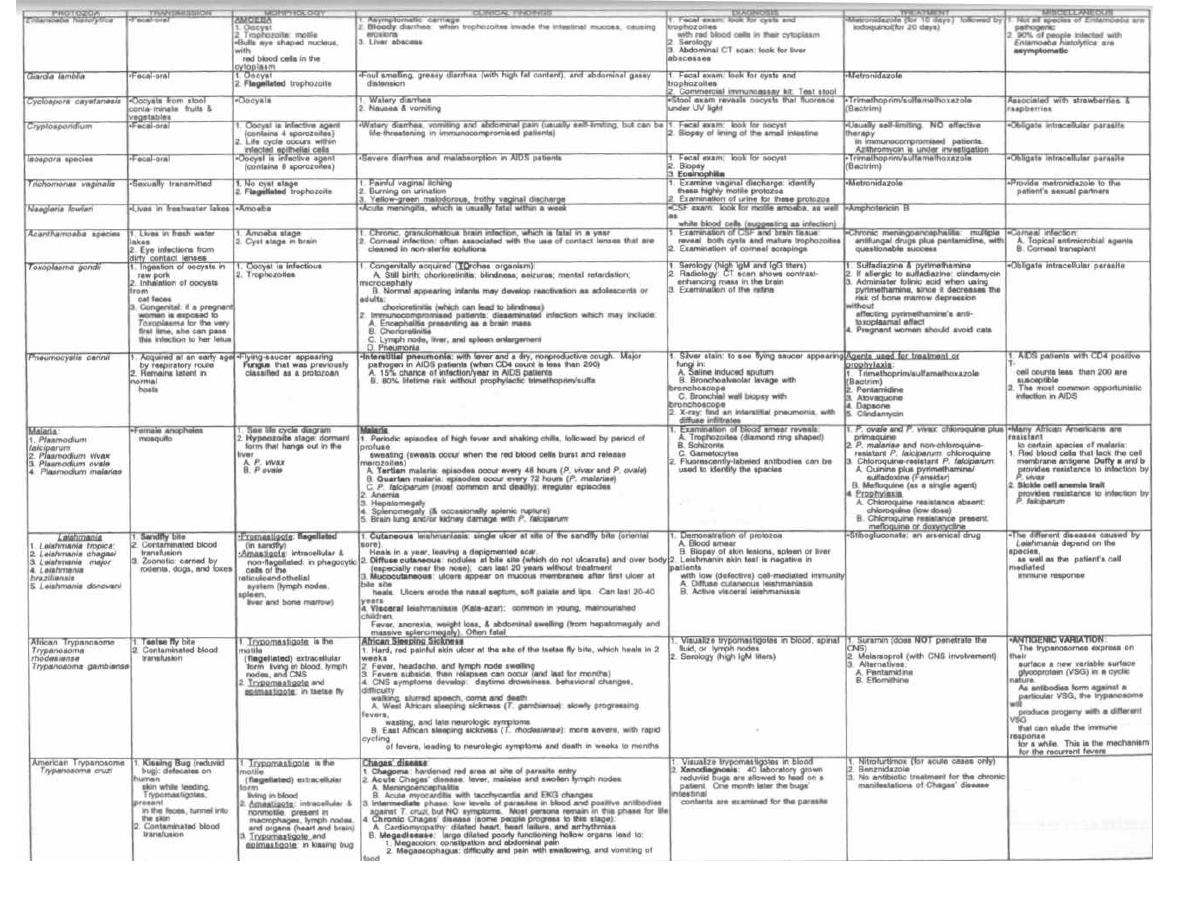
Figure 30-12
PROTOZOA
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple ®MedMaster
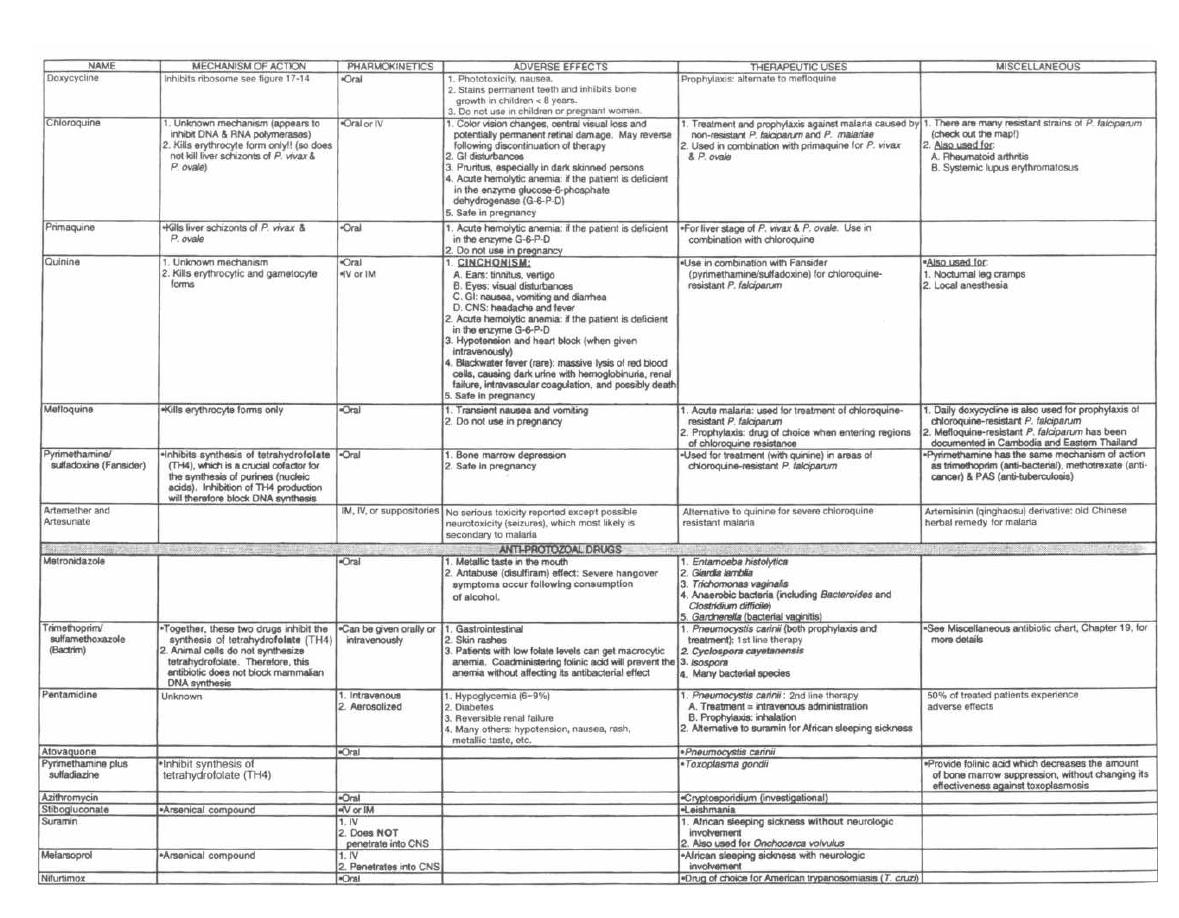
Figure 30-13
ANTI-PARASITIC DRUGS
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple OMedMaster

CHAPTER 31.
Helminths is Greek for "worm." Worms are usually
macroscopic, although diagnosis often requires the vi-
sualization of the eggs, which are microscopic, in the
stool. We will discuss 16 types of worms that cause sig-
nificant infections in humans. The first 10 are round-
worms (nematodes), and the last 6 are the more
primitive flatworms (platyhelminthes). By under-
standing their life cycles, you can learn ways to prevent
or eradicate helminthic infections.
Within the normal human host there is usually
no immune reaction to living worms. However, there
is often a marked response to dead worms or eggs. With
many of the worm infections, our immune system is
kind enough to raise a red flag-elevating the level of
eosinophils in the blood-thereby assisting in diagnosis.
NEMATODES
(Roundworms)
Intestinal Nematodes
"Intestinal" nematodes all mature into adults within
the human intestinal tract. The larval forms of many of
these roundworms may be distributed widely through-
out the body.
Three of the intestinal nematodes are acquired by the
i ngestion of eggs: Ascaris lumbricoides, Trichuris
trichiura ( whipworm), and Enterobius vermicularis
( pinworm). Two worms, Necator americanus ( hook-
worm) and Strongyloides stercoralis, are acquired when
their larvae penetrate though the skin, usually of the
foot. Trichinella spiralis is acquired by the ingestion of
encysted larvae in muscle (pork meat).
With infection, most of these intestinal worms (except
for Enterobius and Trichuris, which stay in the intesti-
nal tract) invade other tissues at some stage of their life
cycle. This stimulates our immune system to raise the
number of eosinophils in the blood.
Fig. 31-1.
The first 3 roundworms (Ascaris lumbri-
coides, Necator americanus, and Strongyloides sterco-
rous)
all have a larval form that migrates through
the tissue and into the lung at some stage of their life
cycle. The larvae grow in the lung, are coughed up and
swallowed into the intestine, where they grow into
adult worms.
1) Ascaris lumbricoides:
Infection occurs in the
tropics and mountainous areas of the southern U. S.,
when individuals consume food that is contaminated
with eggs. Larvae emerge when the eggs reach the
small intestine. The larvae penetrate through the in-
CHAPTER 31. HELMINTHS
251
HELMINTHS
testinal wall and travel in the bloodstream to the lungs.
The larvae grow in lung alveoli until they are coughed
up and swallowed. These larvae again reach the small
i ntestine and mature into adults. Here each adult worm
produces over 200 thousand eggs per day, which are ex-
creted in feces.
2)
Necator americanus:
The larval form lives
in the soil and eats bacteria and vegetation. After
a week it transforms into a long, slender filari-
form larva that can penetrate human skin. The
filariform larva penetrates between the toes of the
hapless human who walks by shoeless. The larvae travel
directly to the alveoli of the lungs, where they grow and
are coughed up and swallowed. The adult worms de-
velop as they arrive at the small intestine, where they
attach by their mouths, and suck blood. At this point,
the hookworms copulate and release fertilized eggs.
3)
Strongyloides stercoralis:
The larval forms in
the soil penetrate the human skin and travel to the lung.
There they grow, are coughed up and swallowed into the
small intestine, where they develop into adult worms
that lay eggs. The eggs are not passed in the stools.
Rather, filariform larvae hatch and can do 3 things:
a) Autoinfection: The filariform larvae pene-
trate the intestine directly, without leaving, and go
to the lung to continue the cycle.
b) Direct cycle: The filariform larvae pass out in
the feces, survive in the soil, penetrate the next
passerby, and migrate to the lungs. This complete
cycle is almost exactly the same as that
of
Necator
americanus ( hookworm).
c) Indirect cycle: This is a sexual cycle where
the filariform larvae are passed out in the stool and
while in the soil develop into male and female
adults. They mate in the soil and produce fertilized
eggs. The filariform larvae then hatch and reinfect a
human, moving to the lung.
Ascaris lumbricoides
Fig. 31-1.
Ascaris infection may be mild or asympto-
matic. With heavy infections the patient may develop ab-
dominal cramping. Severe infections involve adult worm
invasion into the bile ducts, gall bladder, appendix, and
liver. Children with heavy worm loads may suffer from
malnutrition because the worms compete for the same food
and sometimes a mass of worms can actually block the in-
testine. When the larvae migrate into the lung, the patient
may develop cough, pulmonary infiltrate on chest
x-ray, and a high eosinophil count in the blood and sputum.
Diagnosis is made by identification of eggs in feces. A
sputum examination may reveal larvae. The peripheral
blood smear may also reveal an increased number of
eosinophils.
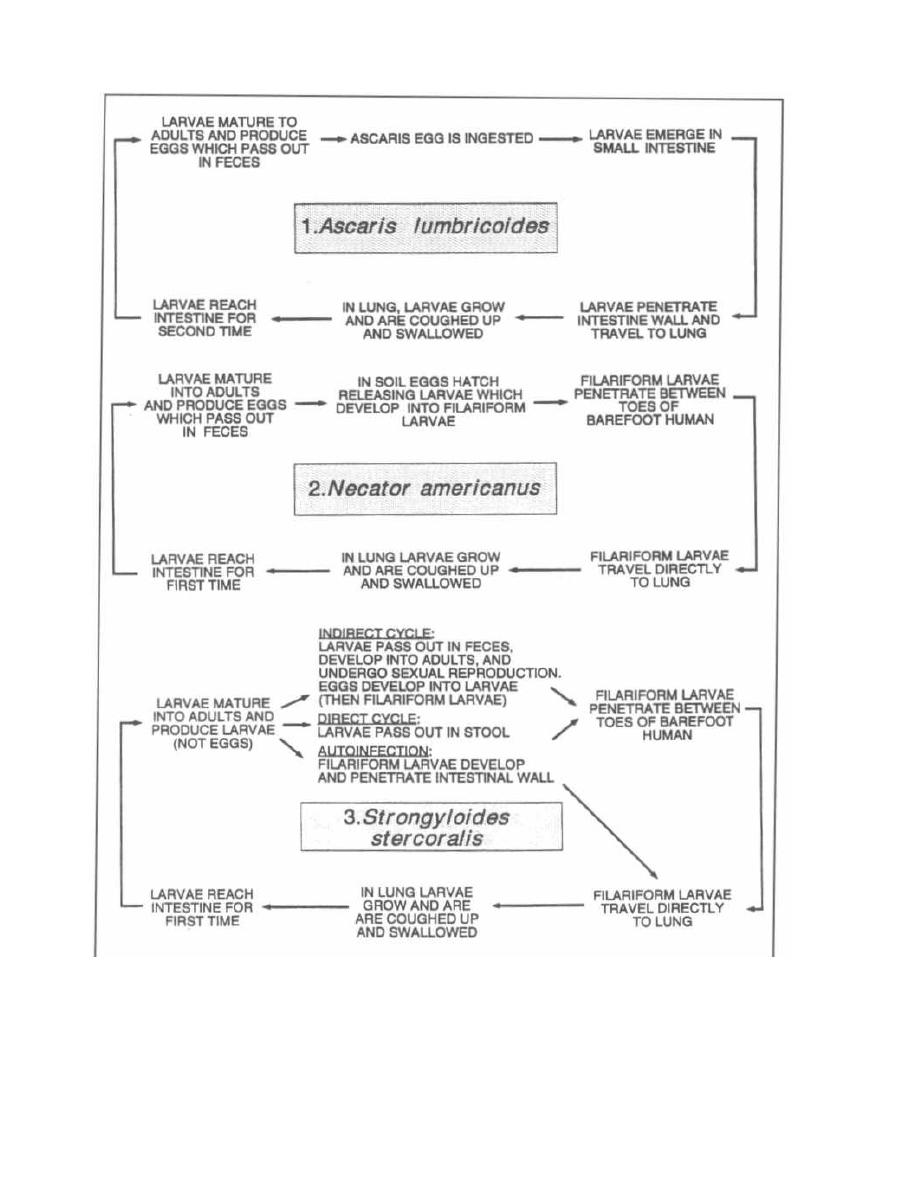
Figure 31-1
Treatment
The intestinal nematodes Ascaris lumbricoides,
Necator Americanus (hookworm), Strongyloides sterco-
CHAPTER 31. HELMINTHS
252
ralis, Enterobius vermicularis (pinworm), Trichuris
trichiura (whipworm), and Trichinella spiralis are all
treated with the same drug:

Think of a worm,
Worms BEND,
BEND them a lot and you can kill them!
Mebendazole
Thiabendazole
Albendazole
These drugs paralyze the roundworms so they are
passed out in the stool. Other drugs will irritate the
worms, causing them to migrate out of the small intes-
tine to other organs, which can be fatal.
Pyrantel pamoate is an alternative agent for En-
terobius (pinworm), Necator (hookworm), and Ascaris.
Necator americanus and Ancyclostoma
duodenale
(Hookworm)
Fig. 31-1. Notice that Ascaris lumbricoides and Neca-
tor americanus (hookworm) have very similar life cy-
cles. They differ only in the path that each larvae form
takes to reach the lung: Necator, foot to lung; Ascaris,
intestine to lung.
The patient with hookworm can develop diarrhea, ab-
dominal pain, and weight loss. Since each hookworm
sucks blood from the wall of the intestine, hookworm in-
fection may cause an iron deficiency anemia. There is
also an intense itching and rash at the site of penetra-
tion through the skin (between the toes), and the local
growth in the lung can result in a cough, infiltrate on
chest x-ray, and eosinophilia.
Diagnosis is made by identifying eggs in a fresh fecal
sample. This infection may be treated with mebenda-
zole. Also treat the iron deficiency anemia.
Strongyloides stercoralis
Fig. 31-1. Individuals infected with Strongyloides
may complain of vomiting, abdominal bloating, diarrhea,
anemia, and weight loss. Similar to hookworm infection,
patients may develop a prurititic rash, lung symptoms
(cough or wheezing), and/or eosinophilia. When patients
infected with strongyloides are given immunosuppres-
sive medications, such as prednisone, they can develop
a severe autoinfection. The filariform larvae will invade
the intestine, lung and other organs, causing pneumonia,
ARDS, and multi-organ failure. Patients with COPD
and asthma living in areas endemic for Strongyloides
should have their stool and eosinophil count checked be-
fore steroid treatment!
Diagnosis is made by identifying larvae in feces (no
eggs). The enterotest, where a long nylon string is
swallowed and later pulled back out the mouth, may
demonstrate larvae of
Strongyloides.
Sputum exam
CHAPTER 31. HELMINTHS
253
may also demonstrate larvae. Examination of the blood
will reveal an elevated level of eosinophils.
Treat infected patients with thiabendazole or al-
bendazole and ivermectin
Trichinella spiralis
You may have seen Gary Larson's "The Far Side" comic
showing wolves at the edge of a pork farm, saying, "Let's
rush them and Trichinella be damned." After reading
about Trichinella spiralis, we can finally get the joke.
Infection occurs following the ingestion of the encysted
larvae of Trichinella spiralis, which are often present in
raw pork. After ingestion, the cysts travel to the small in-
testine, where the larvae leave the cysts and mature into
mating adults. Following mating, the adult males are
passed in the feces. The females penetrate into the in-
testinal mucosa, producing thousands of larvae. The lar-
vae then enter the bloodstream and spread to organs and
skeletal muscle. The larvae then become encysted in
skeletal muscle, where they may last for decades.
Most patients are asymptomatic with the initial
infection. Some patients will complain of abdominal
pain, diarrhea, and fever as the worms mature in the
small intestine and penetrate through the intestinal
wall. About 1 week after the initial intestinal invasion,
the larvae migrate into skeletal muscle, producing
fevers and muscular aches. In severe (sometimes fatal)
cases, larvae may invade heart muscle and brain tissue.
Diagnosis can be made with serologic tests or muscle
biopsy, which will reveal the encysted larvae. Since there
is significant muscle invasion, examination of the differ-
ential white blood cell count will reveal a marked increase
in the percentage of eosinophils. The invasion of muscle
also results in increased levels of serum muscle enzymes,
such as creatinine phosphokinase (CPK).
Prevention is best carried out by killing cysts, either by
cooking or freezing the pork meat prior to consumption.
Mebendazole and thiabendazole have little effect
on muscle larvae but may be helpful.
Trichuris trichiura
(Whipworm)
The next 2 worms, Trichuris trichiura and Enterobius
vermicularis, have very simple life cycles: there is no fi-
lariform larvae stage, no tissue invasion, and no lung in-
volvement. Since there is no tissue invasion, the
eosinophil count is not elevated.
Fig. 31-2.
Trichuris trichiura has a simple slow
life cycle. Like the Congressional whip trying to move
his party to a decision, the whipworm life cycle is
slow. Transmission occurs by ingestion of food conta-
minated with infective eggs. The eggs hatch in the
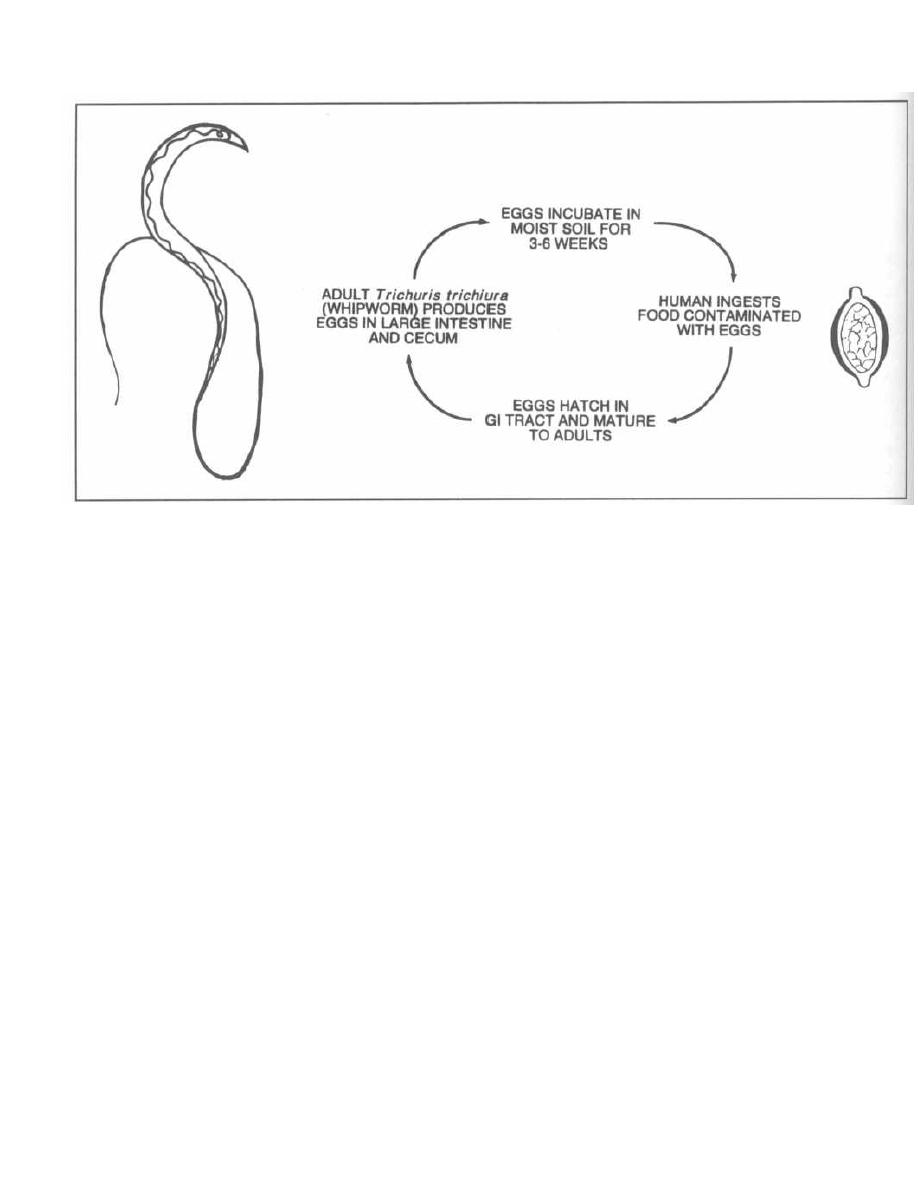
Figure 31-2
gastrointestinal tract and migrate to the cecum and
ascending large intestine. The mature adult will then
produce thousands of eggs per day for about one year.
There is no autoinfection, since the eggs must incu-
bate in moist soil for 3-6 weeks before they become
infective.
Infected patients may have abdominal pain and diar-
rhea. Diagnosis is made by identifying eggs in fecal
specimens. The eggs look like footballs with bumps on
each end. The adults have the appearance of a bullwhip,
thus the common name.
Treat patients with mebendazole.
Enterobius vermicularis
( Pinworm)
The
Enterobius'
life cycle is very simple: The eggs are
simply ingested, and the pinworms mature in the cecum
and ascending large intestine. The female migrates to
the perianal area (usually at night) to lay her eggs,
which become infectious 4-6 hours later. This infection
causes severe perianal itching. An infected individual
will scratch the perianal area and then reinfect himself
or others (hand-to-mouth) because his hands are now
covered with the microscopic pinworm eggs.
This infection is more of a nuisance than it is danger-
ous. The infected individual has a terribly itchy behind,
usually at night.
Diagnosis is made by placing scotch tape firmly on the
perianal area. The scotch tape will pick up eggs, which
CHAPTER 31. HELMINTHS
25 4
can be viewed under a microscope. At night the larger
adult females can sometimes be seen with the unaided
eye, crawling across the perineal area. There is no
eosinophilia, since there is no tissue invasion.
Treat with mebendazole or pyrantel pamoate,
avoid scratching, and change the sheets daily.
Fig. 31-3. Enterobius v
ermicularis . The female worm is
like an intestinal (entero) bus. She drives out of the
anus every night and drops off her egg passengers on
the perineum. If a pin (pinworm) pops her tire, seal
the leak with some scotch tape. The scotch tape test is
used to diagnose
Enterobius vermicularis
i nfection.
Blood and Tissue Nematodes
The blood and tissue nematodes are not spread by fe-
cal contamination, but rather by the bite of an arthro-
pod. These threadlike, round worms belong to the
family Filarioidea and so are called filariae. Adult fi-
lariae live in the lymphatic tissue and give birth to
prelarval forms (they do not lay eggs) called microfi-
lariae. The microfilariae burrow through tissue and
circulate in the blood and lymphatic system. Microfilar-
iae are picked up by bloodsucking arthropods, which
transmit them to another human. Disease is caused
by allergic reactions to both the microfilariae and
dead adult worms in the lymphatic system as well as
other organs (such as the eyes with
Onchocerca volvu-
lus
i nfection).

Figure 31-3
Onchocerca volvulus
This filarial infection is found in Africa and Central
and South America. The larvae are transmitted to hu-
mans by the bite of infected black flies. The microfi-
lariae (larvae) mature into adults, which can be found
coiled up in fibrous nodules in the skin and subcuta-
neous tissue. After mating, the adults produce micro-
filariae, which migrate through the dermis and connec-
tive tissue. Patients will develop a pruritic skin rash
with darkened pigmentation. The skin may thicken
with the formation of papular lesions that are actually
intraepithelial granulomas. The thickened skin may
appear dry, scaly, and thick ("lizard skin"). Microfilar-
iae may also migrate to the eye, causing blindness
(since the black fly vector breeds in rivers and streams,
this is often referred to as "river blindness"). There
are villages in endemic areas where most of the inhabi-
tants are blind.
Diagnosis is made by demonstrating microfilariae in
superficial skin biopsies, or adult worms in a nodule.
Microfilariae can often be seen in the eye (cornea and
anterior chamber) by slit lamp examination.
A new drug, ivermectin, has revolutionized the
treatment of Onchocerciasis. It kills microfilariae and
prevents the microfilariae from leaving the uteri of
adult worms. The manufacturer (Merck) has donated
the drug to the World Health Organization for a pro-
gram to eradicate Onchocerca from the planet. As hu-
CHAPTER 31. HELMINTHS
255
mans are the only reservoir, treating people in endemic
areas for 10 years (as planned) will prevent the birth of
new microfilariae while all the adult worms (which have
long life spans) die of old age.
If you know Spanish, ver means "to see." So: I VER
with IVERmectin!!
Wuchereria bancrofti
and
Brugia malayi
( Elephantiasis)
Wuchereria bancrofti and Brugia malayi both cause a
lymphatic infection that can result in chronic leg
swelling. Wuchereria infection is endemic to the Pacific
Islands and much of Africa, while Brugia is endemic to
the Malay Peninsula and is also seen in much of South-
east Asia. Transmission occurs by the bite of an infected
mosquito. The transmitted microfilariae mature into
adults within the lymphatic vessels and lymph nodes of
the genitals and lower extremities. Mature adults mate,
and their offspring (microfilariae) enter nearby blood
vessels.
Small infections may only result in enlarged lymph
nodes. Frequent infections in endemic areas result in
acute febrile episodes, associated with headaches and
swollen inguinal lymph nodes. Occasionally, following
repeated exposures, fibrous tissue will form around
dead filariae that remain within lymph nodes. The fi-
brous tissue plugs up the lymphatic system, which re-
sults in swelling of the legs and genitals. The swollen

Figure 31-4
(edematous) areas become covered with thick, scaly
skin. This chronic disfiguring manifestation is called
elephantiasis because the extremities take on the ap-
pearance of elephant legs.
Diagnosis is made by the identification of microfilar-
iae in blood drawn at nighttime. (For poorly understood
reasons, very few organisms circulate during daylight
hours. This is called Nocturnal periodicity.) Diagno-
sis can also be made by identification of positive anti-
body titers via immunofluorescence.
Diethylcarbamazine is the agent used to treat ele-
phantiasis. Lymphatic damage may require surgical
correction.
Fig. 31-4. There are two ( Di) women named Ethyl in
this car: Di-ethyl-car. You will notice that there is an
elephant between Ethyl and Ethyl. You see, the drug
Diethylcarbamazine is used to treat the filarial infec-
tions caused by
Wuchereria bancrofti, Brugia malayi,
Loa boa,
and
Onchocerca volvulus.
These filariae chron-
ically infect the lymphatics, causing lymph obstruction,
giant swollen testicles, and elephant legs ("elephantia-
sis"). Thus the elephant between the Ethyls.
Dracunculus medinensis
(Guinea worm)
This very interesting tissue-invasive nematode lives as
a larval form inside intermediate hosts: African and
Asian freshwater copepods (tiny crustaceans). When a
person drinks water containing the microscopic crus-
taceans, the larvae penetrate the intestine and move
deep into subcutaneous tissue, where the adults develop
and then mate. The male is thought to die, but the female
grows over the course of a year to a size of 100 cm!!! She
then migrates to the skin and a loop of her body pokes out
and exposes her uterus. When the uterus comes into con-
tact with water, thousands of motile larvae are released.
Persons infected with
Dracunculus medinensis
will expe-
CHAPTER 31. HELMINTHS
256
rience allergic symptoms, including nausea, vomiting,
hives and breathlessness, during the larval release.
A common practice used to remove the worm involves
driving a small stick under the part of the worm's body
that is looped out of the skin. The stick is slowly twisted
each day to pull out the 100 cm
Dracunculus.
Cutaneous Larval Migrans
Also called creeping eruption, this intensely pru-
ritic, migratory skin infection commonly occurs in the
Southeastern U.S. The larvae of dog and cat hookworms
penetrate the skin and migrate beneath the epidermis.
As these larvae move (a few centimeters per day), an al-
lergic response is mounted, resulting in a raised, red,
itchy rash that moves with the advancing larvae.
The dog hookworm
Ancyclostoma braziliense
and
other species are responsible.
Human tissue-invasive nematodes such as the hook-
worm
(Necator americanus)
and
Strongyloides sterco-
ralis
can produce similar creeping eruptions.
Diagnosis is made by biopsy of the advancing edge of
the rash.
PLATYHELMINTHES
(Flatworms)
Flatworms do not have a digestive tract. There are
2 groups:
1) Trematodes, also known as flukes, include the
freshwater-dwelling schistosomes. Both male and fe-
male members exist and mate within humans (not in
the digestive tract, however). All flukes have a water
snail species as an intermediate host.
2) Cestodes, also known as tapeworms, live and
mate within the human digestive tract. Each tapeworm
has both male and female sex organs (hermaphrodites),
so individual tape worms can produce offspring by
themselves.
Trematodes
(Flukes)
Schistosoma
(Blood Flukes)
Schistosomal infections are extremely common
worldwide, second only to malaria as a cause of sickness
in the tropics. Schistosomes are found in freshwater.
They penetrate through exposed skin and invade the ve-
nous system, where they mate and lay eggs. Since the
eggs must reach freshwater to hatch, schistosomes can-
not multiply in humans.
Fig. 31-5. The 3 major
Schistosoma
species worldwide.

Figure 31-5
SCHISTOSOMES
The
Schistosoma
life cycle begins when their eggs
hatch in freshwater. Larvae emerge and swim until
they find a freshwater snail. After maturing within
these snails, the larvae are released and are now infec-
tious to humans. Mature schistosomal larvae (called
cercariae, which look like little tadpoles with oral
suckers on one end and a tail on the other), penetrate
through exposed human skin, and travel to the intra-
hepatic portion of the portal venous system. At this lo-
cation, the schistosomes rna`ture and mate. Depending
on the species, the pair of schistosomes will migrate to
the veins surrounding either the intestine or the blad-
der, where they lay their eggs. These eggs may enter the
lumen of the intestine or bladder, where they may be ex-
creted via feces or urine into a nearby stream or lake so
that they can continue their life cycle.
Interestingly, the adult worms in the venous system
are able to survive and release eggs for years, since they
are not killed by the immune system. It is thought that
schistosomes practice molecular mimicry (incorpora-
tion of host antigens onto their surface, which fools the
host's immune system into thinking that the schisto-
somes are NOT foreign).
However, cercariae (mature larvae) and eggs briskly
stimulate the immune system, and are responsible for
the systemic illness caused by this infection.
Clinical Manifestations
Schistosomiasis has 3 major disease syndromes that
occur sequentially: 1) Dermatitis as the cercariae pene-
trate a swimmer's skin, 2) Katayama fever as the
grown adults begin to lay eggs, and 3) Chronic fibrosis
of organs and blood vessels from chronic inflammation
around deposited eggs.
Upon penetration through the skin, patients tran-
siently develop intensely itchy skin (swimmer's itch)
and rash. Katayama fever follows 4-8 weeks later
with symptoms that can include fever, hives, headache,
weight loss, and cough. These symptoms may persist for
3 weeks. Lymph node, liver, and spleen enlargement
with eosinophilia are common.
When the schistosomes set up their home in the veins
surrounding the intestines or bladder, they begin re-
leasing eggs, many of which do not reach the feces or
CHAPTER 31. HELMINTHS
25 7
urine. Instead, these eggs are whisked off by the blood-
stream, where they are deposited in the liver, lung, or
brain. The immune system reacts against these eggs,
walling them off as granulomas. The deposition of eggs
in the venous walls of the liver leads to fibrosis, which
causes blockage of the portal venous system and a
backup of venous pressure into the spleen and mesen-
teric veins (portal hypertension). Any blood vessels or
organs that these eggs get stuck in can become in-
flamed, with formation of granulomas and ulcers. Pa-
tients may develop hematuria, chronic abdominal pain
and diarrhea, brain or spinal cord injury, or pulmonary
artery hypertension.
Diagnosis is based on the demonstration of eggs in
stool or urine samples and high eosinophil counts in the
blood. Control is directed at the proper disposal of hu-
man fecal waste and elimination of the snails that act
as the intermediate host.
Treatment
A group of quantum physicists got together to eradi-
cate this horrible disease. They wanted a drug that kills
all the flukes and tapeworms also. They succeeded so
Praise the quantum physicists! ( Note: This is a lie).
Praziquantel: This is truly a broad spectrum anti-
helminthic agent covering cestodes and trematodes
alike. When treating patients with praziquantel, don't
be surprised if there is an immediate exacerbation of
symptoms. The death of these schistosomes evokes a
vigorous immune response.
Cestodes
(Tapeworms)
Tapeworms are flatworms that live in the intestine of
their host. Since they lack a true digestive tract, they
suck up nutrients that have already been digested by
their host. Tapeworms are hermaphrodites (both male
and female organs in the same tapeworm), which en-
ables a single tapeworm to produce offspring. Humans
are usually the host of the adult tapeworm while other
animals may serve as intermediate hosts for the larval
stages.
Species
Geographic distribution
Resides in veins
surrounding
Deposits eggs
in:
Schistosoma japonicum
Eastern Asia
I ntestinal tract
Feces
Schistosoma mansoni
South America and Africa Intestinal tract
Feces
Schistosoma
haematobium
Africa
Bladder
Urine
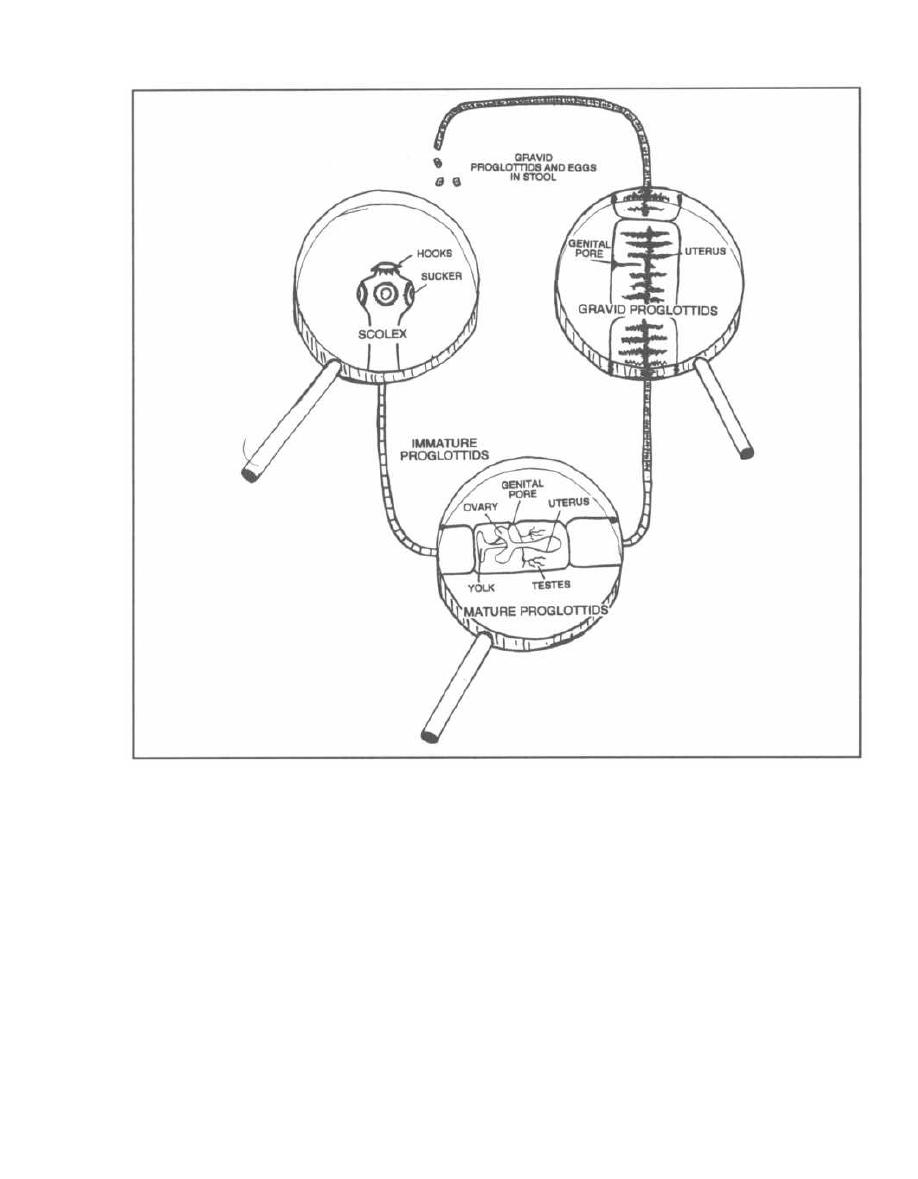
CHAPTER 31. HELMINTHS
Figure 31-6
Fig. 31-6. The tapeworm is long and flat (like a
typewriter ribbon) and consists of a chain of boxlike seg-
ments called proglottids. Let us examine the tape-
worm from its head down:
1) Scolex: The most anterior segment of the tape-
worm (the head), which has suckers and some-times
hooks.
2) Immature proglottids.
3) Mature proglottids have both male and female sex
organs.
4) Gravid proglottids contain fertilized eggs.
All of the tapeworm infections can be treated with
praziquantel or niclosamide.
258
Fig. 31-7. Niclosamide is an alternative agent to
praziquantel for treatment of tapeworm (cestode) infec-
tions. Picture a tapeworm wrapped around a nickel or
a nickel under tape.
Albendazole and praziquantel are used for the treat-
ment of
Taenia solium
larval cysticerci (see below).
Taenia solium
( Pork Tapeworm)
Humans acquire this infection by ingestion of under-
cooked pork infected with larvae. The pork tapeworm
attaches to the mucosa of the intestine via hooks on its
scolex and grows to a length of 2-8 meters. It releases
Figure 31-7
eggs in the human feces. When pigs graze on fields con-
taminated with the egg-infested human feces, they be-
come the intermediate host. The eggs develop into
larvae that disseminate through the intestine and into
muscle. In the animal's muscle tissue, the larvae de-
velop into another larval form, the cysticercus. The
cysticercus is a round, fluid-filled bladder with the lar-
val form within. When a human eats insufficiently
cooked pork muscle, the larval cysticercus converts to
the adult tapeworm in the intestine, and the cycle con-
tinues (see Fig. 31-8).
Infected individuals usually have minimal symp-
toms. Diagnosis is made by demonstration of proglottids
and/or eggs in a fecal sample.
Cysticercosis occurs when humans play the role of
the pig and ingest eggs rather than encysted lar-
vae. These eggs hatch within the small intestine, and
the larvae migrate throughout the body, where they
penetrate into tissue and encyst, forming cysticerci in
the human. Most commonly, the larvae encyst in the
brain and skeletal muscles (see Fig. 31-8).
CHAPTER 31. HELMINTHS
259
Cysticerci in the brain tend to cause more symptoms,
and this condition is called
Neurocysticercosis.
There are
usually 7-10 cysts in the brain, causing seizures, ob-
structive hydrocephalus, or focal neurologic deficits. The
cysts grow slowly and after 5-10 years begin to die and
leak their fluid contents. The antigenic contents cause lo-
cal inflammation and enhanced symptoms (more
seizures, meningitis, hydrocephalus, and focal deficits).
In endemic areas such as Mexico, Central and South
America, the Philippines, and SE Asia, Cysticercosis is
the most common cause of seizures.
The diagnosis of cysticercosis is made with the help of
a CAT scan or biopsy of infected tissue (brain or mus-
cle), both of which may reveal calcified cysticerci. Newer
serologic tests are also proving helpful in the diagnosis
of cysticercosis. Symptomatic disease, especially neuro-
cysticercosis, can be treated medically with dibendazole
or praziquantel.
Note that our immune system raises a red flag to
mark this invasion: the elevation of the eosinophil level
in the blood.
Taenia saginata
( Beef Tapeworm)
Taenia saginata
has the exact same life cycle as does
Taenia solium,
except that humans do not develop cys-
ticerci when they ingest eggs, as do the intermediate
hosts (cattle). For this reason, beef tapeworm infection
is relatively benign.
The beef tapeworm is acquired by the ingestion of lar-
val cysticerci in undercooked beef muscle. The adult
beef tapeworm develops and adheres (via suckers on its
scolex) to the intestinal mucosa, where it may reach a
length of over 10 meters and contain more than 2 thou-
sand proglottids.
Patients are usually asymptomatic, although they
may develop malnutrition and weight loss. Diagnosis
is made by identifying gravid proglottids and/or eggs
in feces.
Diphyllobothrium latum
(Fish Tapeworm)
The fish tapeworm can grow to 45 meters in length.
It is acquired by ingesting larvae in raw freshwater
fish. The life cycle involves the human and 2 fresh-
water intermediate hosts, a crustacean and a fish. The
adult tapeworms in the human intestine drop off their
gravid proglottids loaded with eggs. When the eggs
end up in water, they convert to a motile larval form,
which is then ingested by a crustacean, which is then
i ngested by a freshwater fish (trout, salmon, pike, etc.),
which is then ingested by a human-to end this long
sentence!
CYSTICERCI
LARVAE DEVELOP
INTO ADULT
TAPEWORMS IN
HUMAN INTESTINE
GRAVID
PROGLOTTIDS
AND EGGS
PASS IN FECES
HUMAN
HUMAN EATS
PORK WITH
u
CYSTICERCI
CYSTICERCOSIS:
HUMAN INGESTS EGGS.
HATCHED LARVAE
PENETRATE HUMAN MUSCLE,
BRAIN, LIVER, HEART, AND FORM
CYSTICERCI
PIGS INGEST
EGGS
PIG
CYSTICERCI
DEVELOP IN
PIG MUSCLE
1
LARVAE DEVELOP
AND MIGRATE INTO
PIG MUSCLE
Figure 31-8.
Pork tapeworm life cycle.
The large intestinal
Diphyllobothrium latum
tape-
worm provokes few abdominal symptoms, although it can
absorb vitamin
B12
to a significant degree. If vitamin
B,2
deficiency develops, anemia will occur. Diagnosis of in-
fection is made by identification of eggs in the feces.
Hymenolepis nana
(Dwarf Tapeworm)
This is the smallest tapeworm that infects humans
(only 15-50 mm in length), and it has the simplest life cy-
cle. There are NO intermediate hosts. Humans ingest
eggs that grow into adult tapeworms, and the adult tape-
worms pass more eggs that are again ingested by humans.
An infected patient will complain of abdominal
discomfort and occasionally nausea and vomiting.
Diagnosis is made by demonstration of eggs in a fecal
sample.
Echinococcus granulosus and
multilocularis
(Hydatid Disease, an Extra-intestinal Tapeworm
Infection)
Fig. 31-9. Dogs and sheep perpetuate the life cycle of
Echinococcusgranulosus
and the human is only a dead-
CHAPTER 31. HELMINTHS
260
end in the cycle.
Echinococcus
shares many similarities
with
Taenia solium,
with humans ingesting the eggs.
These eggs hatch in the intestine and develop into lar-
vae. After penetrating through the intestinal wall, the
larvae disseminate throughout the body. Most larvae
are concentrated in the liver, but larvae may also infect
the lungs, kidney and brain.
Each larva forms a single, round fluid-filled "hydatid"
cyst. These hydatid cysts can undergo asexual budding
to form daughter cysts and protoscolices inside the orig-
inal cyst. They can grow to 5-10 cm in size. Each cyst
may cause symptoms because it compresses the organ
around it (in the liver, lung, or brain). Humans are ex-
tremely allergic to the fluid within the cyst, and if the
cyst bursts, the allergic reaction may be fatal. While the
cysts of
Echinococcus granulosus
simply grow larger,
only to spread if they rupture, E.
multilocularis
cysts
undergo lateral budding and spread. These spreading
cysts can be misdiagnosed as a slowly growing tumor.
Diagnosis of the hydatid cyst employs similar tech-
niques as with
Taenia solium's
cysticerci cysts, using
CAT scanning and tissue biopsy. Only 10% of hydatid
cysts cause symptoms, and treatment of these is diffi-
cult. The best thing to do is to cut them out of the liver,
lung, or (yikes!) brain, but if the cyst fluid spills, out
will pour daughter cysts, protoscolices, and highly
EGGS IN DOG
'
SHEEP INGEST
STOOL
EGGS
LARVAE MIGRATE
INTO TISSUE
TAPEWORM
SHEEP
HYDATID CYSTS
FORM IN TISSUE
USUALLY
ONLY A SINGLE
HYDATID CYST
FORMS IN TISSUE
TISSUE WITH
CYSTS RUPTURE
CYST CAUSES
TAPEWORMS
INTO TISSUE
DAMAGE TO
INGESTED BY DOG
RELEASE TAPEWORMS
ORGANS
Figure 31-9.
Echinococcus life cycle.
allergenic fluid. Optional treatment usually starts with
treatment for months with albendazole to kill the cysts
(although this alone is rarely curative). The cyst is then
exposed surgically and some of the cyst fluid is carefully
aspirated. Saline, iodophors, or ethanol is next instilled
into the cyst to make sure the contents are dead. After
about 30 minutes the cyst is cut out.
When a hydatid cyst is inoperable due to a tricky lo-
cation or a poor surgical candidate, therapy with alber-
dazole is initiated and in some centers this is followed
by CAT scan guided fine needle injection of ethanol into
the cyst.
Since dogs and sheep perpetuate the cycle, efforts to-
ward control should target these animals. Dogs in graz-
ing countries should not be fed uncooked animal meat
and should be treated periodically with niclosamide.
Fig. 31-10.
Summary of the helminths.
Fig. 31-11.
Summary of the anti-helminths drugs.
26 1
References
Grove DI. Tissue Nematodes (Trichinosis, Dracunculiasis, Fi-
lariasis. In: Mandell GL, Bennett JE, Dolin R. editors. Prin-
ciples and Practice of Infectious Diseases. 4th edition. New
York: Churchill Livingstone 1995;2531-2537.
King HK. Cestodes (tapeworms). In: Mandell GL, Bennett JE,
Dolin R. editors. Principles and Practice of Infectious Dis-
eases. 4th edition. New York: Churchill Livingstone
1995;2544-2553.
Liu LX, Weller PF. Drug therapy: Antiparasitic drugs. N Engl
J Med 1996;334:1178-84.
Mahmoud AA. Trematodes (Schistosomiasis) and other
flukes. In: Mandell GL, Bennett JE, Dolin R. editors. Prin-
ciples and Practice of Infectious Diseases. 4th edition. New
York: Churchill Livingstone 1995;2538-2544.
Rosenblatt JE. Antiparasitic Agents. Symposium On Antimi-
crobial Agents-Part VII. Mayo Clin Proc 67:276-287, 1992.
Sanford JP, Gilbert DN, et al. Guide To Antimicrobial Therapy
1996. Antimicrobial Therapy, Inc, Dallas Texas; 1996.
CHAPTER 31. HELMINTHS
_
HUMANS INGEST
LARVAE MIGRATE
EGGS
INTO TISSUE
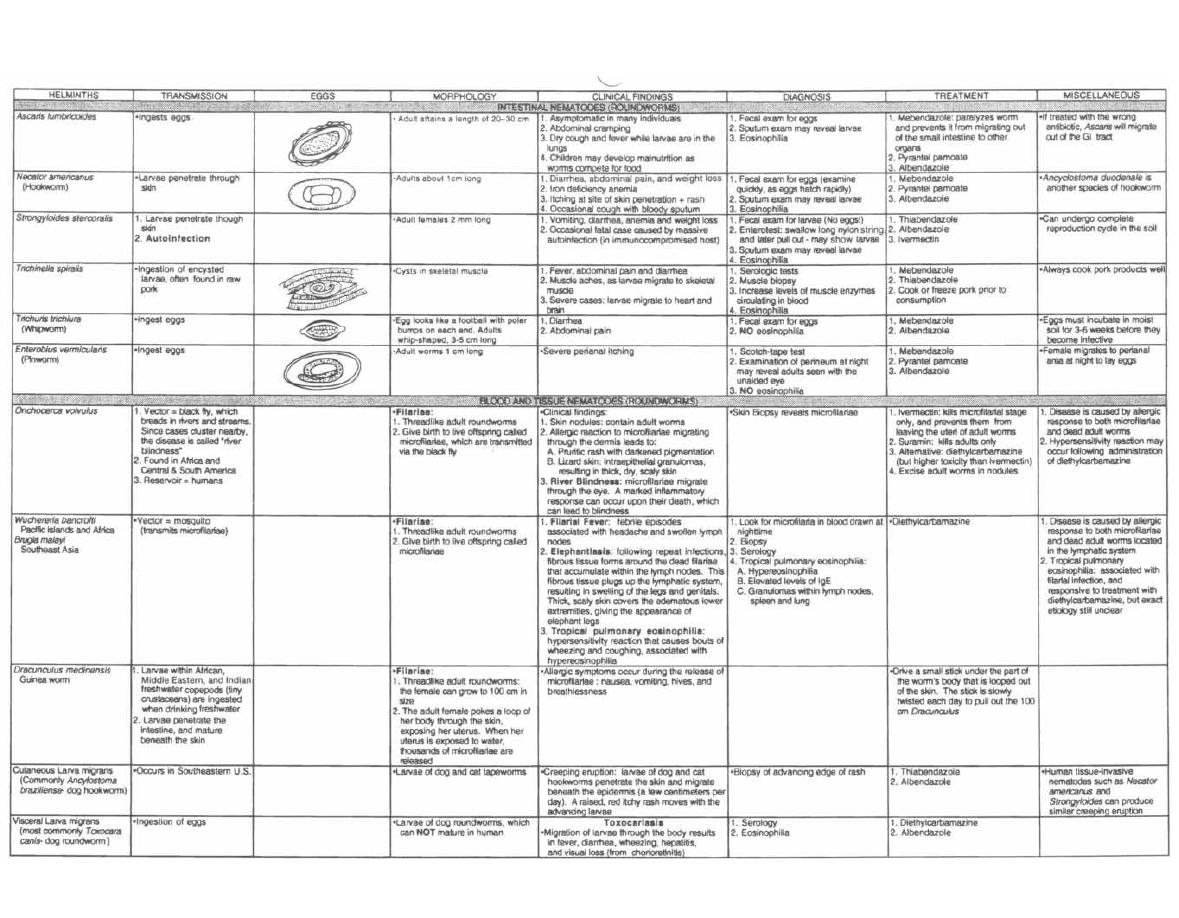
Figure 31-10 HELMINTHS
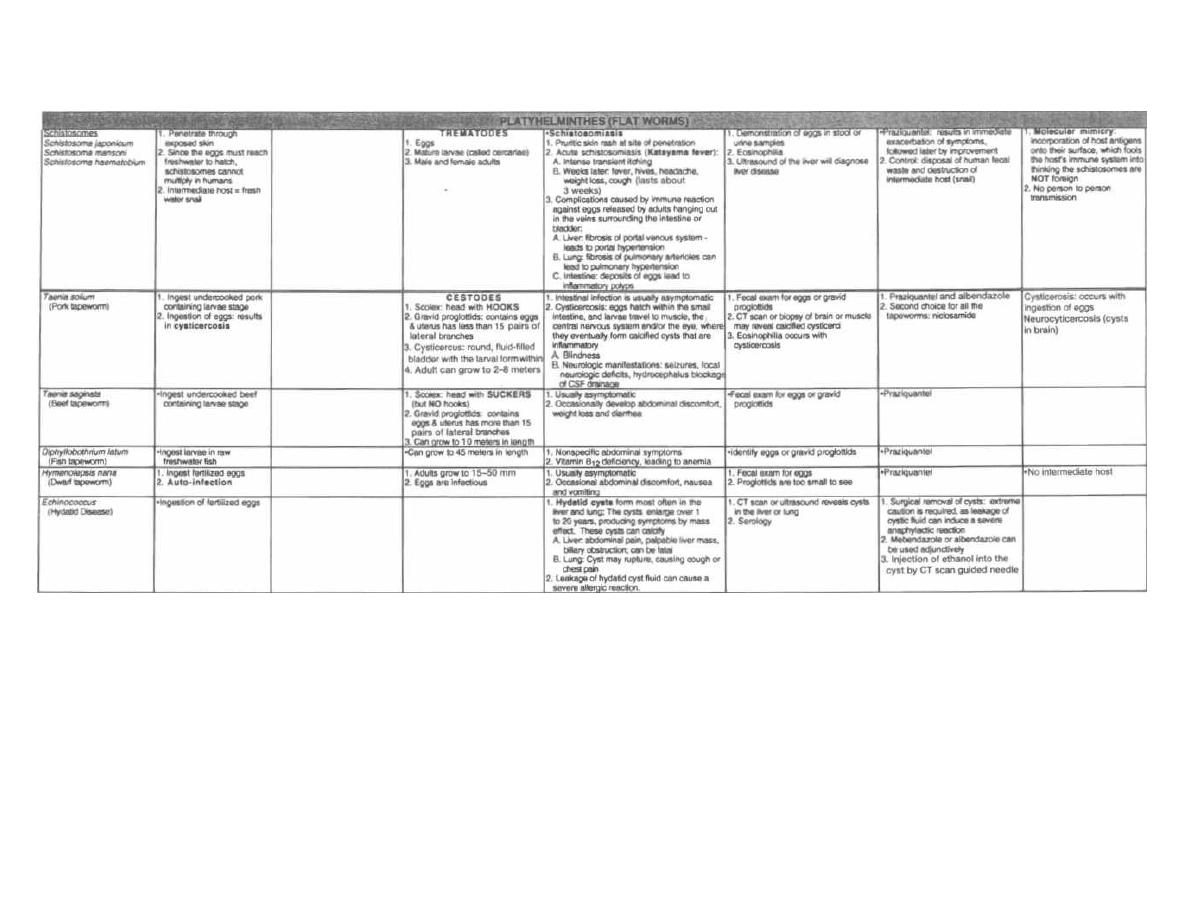
Figure 31-10 (continued)
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
OMedMaster
Figure 31-11
ANTI-HELMINTHS DRUGS
M. Gladwin and B. Trattler, Clinical
Microbiology Made Ridiculously Simple
©MedMaste

Protozoa are free-living, single celled, eucaryotic cells
with a cytoplasmic membrane and cellular organelles,
i ncluding 1 or 2 nuclei, mitochondria, food vacuoles, and
endoplasmic reticulum. They come in many sizes, from
5 micrometers to 2 millimeters. They have an outer
l ayer of cytoplasm (ectoplasm) and an inner layer (en-
doplasm), which appear different from each other under
the microscope.
The protozoa ingest solid pieces of food through a
small mouth called the cytostome. For example, amoe-
bas ( Entamoeba histolitica) can ingest human red blood
cells into their cytoplasm. The protozoa reproduce asex-
ually, undergoing DNA replication followed by division
i nto 2 cells. They also reproduce sexually by the fusion
of 2 cells, followed by the exchange of DNA and separa-
tion into 2 cells again.
When exposed to new environments (such as temper-
ature changes, transit down the intestinal tract, or
chemical agents), the protozoa can secrete a protective
coat and shrink into a round armored form, called the
cyst. It is this cyst form that is infective when ingested
by humans. Following ingestion it converts back into
the motile form, called the trophozoite.
THE INTESTINAL PROTOZOA
There are 5 intestinal protozoa that cause diarrhea.
Entamoeba histolytica causes a bloody diarrhea, and Gi-
ardia lamblia and Cyclospora cayetanensis cause a
non-bloody diarrhea. Both occur in normal individuals.
Cryptosporidium and Isospora belli cause severe diar-
rhea in individuals with defective immune systems
(such as patients with AIDS).
Entamoeba histolytica
This organism is the classic amoeba we have all
heard about. It moves by extending creeping projec-
tions of cytoplasm, called pseudopodia (false feet). The
pseudopodia pull it along or surround food particles.
About 10% of the world population and 1-5% of the U.
S. population are infected with Entamoeba histolytica.
Most of these infections are asymptomatic, as the amoe-
bas live in peace inside their host carriers. These carri-
ers pass the infective form, the cyst, to other individuals
by way of the fecal-oral route. It is noteworthy that ho-
mosexual men commonly are asymptomatic carriers.
Fig. 30-1.
The motile feeding form of the amoeba is
the trophozoite, which cruises along the intestinal wall
CHAPTER 30. PROTOZOA
PART 4. PARASITES
CHAPTER 30. PROTOZOA
234
eating bacteria, other protozoa, and even human in-
testinal and red blood cells. This trophozoite can convert
to a precyst form, with two nuclei, that matures into a
tetranucleated cyst as it travels down and out the colon.
The precyst contains aggregates of ribosomes, called
chromotoid bodies, as well as food vacuoles that are
extruded as the cell shrinks to the mature cyst; it is the
mature cyst that is eaten, infecting others.
Sometimes (10% of infected individuals) the tropho-
zoites invade the intestinal mucosa causing erosions.
This results in abdominal pain, a couple of loose stools
a day, and flecks of blood and mucus in the stool. The in-
fection may become severe, with bloody, voluminous di-
arrhea.
The trophozoites may penetrate the portal blood cir-
culation, forming abscesses in the liver, followed by
spread through the diaphragm into the lung. Here the
trophozoite infection causes pulmonary abscesses and
often death (worldwide: 100,000 deaths annually).
The stool is examined for the presence of cysts or
trophozoites. Trophozoites with red blood cells in the
cytoplasm suggest active disease, while cysts or tropho-
zoites without internalized red cells suggest asympto-
matic carriage. CAT scan or ultrasound imaging of the
liver will reveal abscesses if present.
Prevention rests on good sanitation: proper disposal
of sewage and purification (boiling) of water.
Fig. 30-2.
The Metro (metronidazole) runs over
Entamoeba histolytica, Giardia lamblia, Trichomonas
vaginalis, and the anaerobic cocci and bacilli including
Bacteroides fragilis, Clostridium difficile and Gard-
nerella
vaginalis.
This drug is also called Flagyl (its
trade name) because it kills the flagellated bugs, Giar-
dia and Trichomonas.
Adverse effects of metronidazole
There is no drinking allowed on the train because it
travels rapidly and jarringly, causing stomach upset to
passengers that consume alcohol (Antabuse-disulfiram
effect). If you eat the train, as King Kong once attempted,
you would end up with a metallic taste in your mouth.
Giardia lamblia
Fig. 30-1.
Giardia lamblia exists in 2 forms: as a cyst
and as a mature, motile trophozoite that looks like a
kite.
NUCLEI
a
BINUCLEATED PRECYST
NORMAL HOST
Entamoeba histolytica
TROPHOZOITE
GLYCOGEN
VACUOLE
CHROMATOID
BODIES
TETRANUCLEATED CYST
4 NUCLEI
Giardia lamblia
TROPHOZOITE
Figure 30-1
It is estimated that 5% of U.S. adults harbor this or-
ganism, mostly asymptomatically. Outbreaks occur
when sewage contaminates drinking water. The organ-
ism is also harbored by many rodents and beavers;
campers frequently develop
Giardia lamblia
infection
after drinking from "clear" mountain streams.
After ingestion of the cyst,
Giardia lamblia
converts
to the trophozoite form and cruises down and adheres
to the small intestinal wall. The organism coats the
small intestine, interfering with intestinal fat absorp-
tion. The stools are therefore packed with fat, which has
a horrific odor! The patient will have a greasy, frothy
diarrhea, along with abdominal gassy distension and
cramps. Since
Giardia
do NOT invade the intestinal
wall, there is NO blood in the stool!!!
For diagnosis and control of
Giardia:
1) Examination of stool for cysts or trophozoites.
2) Commercial immunoassay kit to detect
Giardia
lamblia
antigens in aqueous extracts of stool specimens.
3) Sanitation measures.
Treat these patients with metronidazole (see Fig.
30-2).
Cryptosporidium
It is now apparent that this critter is everywhere!
Animals and humans are equally infected and about
CHAPTER 30. PROTOZOA
23 5
25% of Americans show serologic evidence of previous
infection. It can cause outbreaks of diarrhea from
contaminated municipal water sources and in infants
in day care centers. Sporadic cases can occur in
travelers.
Cryptosporidium
is ingested as a round oocyst that
contains 4 motile sporozoites. Its life cycle occurs
within the intestinal epithelial cells, and it causes di-
arrhea and abdominal pain. These symptoms are self-
li miting in immunocompetent individuals. However,
in immunocompromised patients (AIDS patients, can-
cer patients, or organ transplant recipients who are
receiving immunosuppressive therapy), this organism
causes a severe, protracted diarrhea that is life-
threatening. These patients may have 3-17 liters of
stool per day.
Currently, there is no effective therapy. A new
macrolide drug, azithromycin (see Chapter 17), is be-
ing studied.
Isospora and Microsporidia
These organisms cause a severe diarrhea in immuno-
compromised individuals. They are transmitted via the
fecal-oral
route. Fortunately, the combination of
trimethoprim with sulfamethoxazole (see Chapter
19) is effective against
Isospora,
while albendazole
(see Chapter 31) can treat
Microsporidia.

Figure 30-2
THE SEXUALLY TRANSMITTED
PROTOZOAN
Trichomonas vaginalis
Fig. 30-3.
Trichomonas vaginalis
is transmitted sex-
ually and hangs out in the female vagina and male ure-
thra. The trophozoite of
Trichomonas vaginalis
is a
flagellated protozoon (as is
Giardia lamblia).
A female patient with this infection may complain of
itching (pruritus), burning on urination, and copious
vaginal secretions. On speculum examination you will
CHAPTER 30. PROTOZOA
236
find a thin, watery, frothy, malodorous discharge in the
vaginal vault. Males are usually asymptomatic.
Diagnosis of
Trichomonas:
1) Microscopic examination of vaginal discharge on a
wet mount preparation will reveal this highly motile
parasite.
2) Examination of urine may also reveal
Tri-
chomonas vaginalis.
Treat your patient with metronidazole (see Fig. 30-
2). Provide enough for sexual partners. Even though
males are usually asymptomatic, they must be treated

J
Figure 30-3
or the female partner will be reinfected (since this or-
ganism is not invasive, no immunity is acquired).
THE FREE-L WING MENINGITIS-
CAUSING AMOEBAS
Both
Naegleria fowleri
and
Acanthamoeba
are free-
living amoeba that live in fresh water and moist soils.
Infection often occurs during the summer months when
people swim in freshwater lakes and swimming pools
that harbor these organisms. Although large numbers
of persons are exposed, actual infection rarely occurs.
When it does, the organisms penetrate the nasal mu-
cosa, through the cribriform plate, into the brain and
spinal fluid. Both amoeba can cause an infection of the
meninges and brain (meningoencephalitis).
Naegleria
fowleri
will cause a sudden deadly infection in immuno-
competent persons, while
Acanthamoeba
will cause a
slow granulomatous infection, usually in immunocom-
promised persons.
Naegleria fowleri
Fig. 30-4.
Naegleria
fowleri is known for FOWL
PLAY, since 95% of patients will die within 1 week. In-
fected persons will present with a fever, headache, stiff
neck, nausea, and vomiting, which is very similar to a
bacterial meningitis. If asked, they will give a history of
swimming a week earlier. Examination of cerebrospinal
fluid (CSF) reveals a high neutrophil count, low glucose,
and high protein, exactly like a bacterial meningitis!!!
The Gram stain and culture will reveal NO bacteria,
CHAPTER 30. PROTOZOA
237
and microscopic examination may show the motile
amoeba.
Two patients who survived were treated with in-
trathecal amphotericin B, an antifungal agent.
Acanthamoeba
Acanthamoeba
is responsible for a chronic, granulo-
matous, brain infection in immunocompromised pa-
tients, such as those with AIDS. Over a period of weeks,
they will develop headache, fever, seizures, and focal
neurologic signs. Examination of the CSF and brain tis-
sue will reveal
Acanthamoeba
in both the cyst stage and
trophozoite stage. Treatment is difficult and involves
multiple antifungal drugs with pentamidine.
This organism may also infect the cornea (in im-
munocompetent persons), often when contact lenses are
not properly cleaned. This corneal infection (keratitis)
can lead to blindness. Treatment is with antimicrobial
eye drops.
Fig. 30-5. Comparison of Naegleria and Acan-
thamoeba infection.
THE MAJOR PROTOZOAN
INFECTIONS IN AIDS PATIENTS
The ineffective immune system in AIDS patients sets
them up for certain infections that seldom affect the im-
munocompetent host. We have already discussed 2 par-
asites that can establish a severe, chronic diarrhea in
AIDS patients:
Cryptosporidium
and
Isospora. Two

Figure 30-4
Figure 30-5
THE AMOEBAS
other parasites found more commonly in AIDS patients
than in the general population are Toxoplasma gondii
and Pneumocystis carinii. These protozoa are harbored
by most persons without problems. In AIDS, when the
T-helper cell count drops below 200, these bugs flourish
and cause disease.
Toxoplasma gondii
Many animals are infected with Toxoplasma, and hu-
mans are infected by the ingestion of cysts in under-
cooked meats (raw pork) or food contaminated with
CHAPTER 30. PROTOZOA
238
household cat feces. Kitty litter boxes are the most com-
mon source of exposure for humans, as up to 80% of cats
are infected in the United States. Toxoplasma gondii
undergoes sexual division in the cat and is excreted in
the feces as the infectious cyst.
The protozoan causes disease by reactivation of a la-
tent infection in an immunocompromised person or as a
primary infection in a pregnant woman (leading to
transplacental infection of the fetus).
1) Immunocompromised patients with AIDS or those
who are taking immunosuppressive drugs (for cancer or
Naegleria
Acanthamoeba
A
CUTE meningoencephalitis in normal
hosts
C
HRONIC meningoencephalitis in
immunocompromised hosts
M
ature amoeba only in brain tissue
(NO cysts)
C
ysts and mature amoeba found in
brain tissue
N
o corneal infection
C
orneal infection: diagnose by corneal
scraping

organ transplantation) are susceptible to growth of the
latent
Toxoplasma gondii.
The infection can present in
many ways-with fever; lymph node, liver, and spleen
enlargement; pneumonia; or frequently with infection of
the meninges or brain. In fact,
Toxoplasma
encephalitis
is the most common central nervous system infection
in AIDS patients. The brain infection can involve a
growing mass, much like a tumor, with symptoms of
headache and focal neurologic signs (seizures, gait in-
stability, weakness, or sensory losses). Infection of the
retina, chorioretinitis, is also common, resulting in vi-
sual loss. Examination of the retina reveals yellow-
white, fluffy (like cotton) patches that stand out from
the surrounding red retina.
2) Toxoplasma
is one of the transplacentally ac-
quired TORCHES organisms that can cross the blood-
placenta barrier (see Fig. 26-2.). Transplacental fe-
tal infection can occur if a pregnant woman who has
never been previously exposed to
Toxoplasma gondii
is infected. Congenital toxoplasmosis does not occur
in pregnant women who have serologic evidence of
previous exposure, most likely because of a protec-
tive immune response. Pregnant women should
avoid cats!!
Like rubella (see Chapter 28), congenital toxoplasmo-
sis can cause many problems, including chorioretinitis,
blindness, seizures, mental retardation, microcephaly,
encephalitis, and other defects. If the infection is ac-
quired early during gestation, the disease is severe, of-
ten resulting in stillbirth. Interestingly, infants that
appear normal can develop disease later in life. Clinical
reactivation results most commonly in retinal inflam-
mation (chorioretinitis, which can result in blindness)
that flares late in life (peak incidence in second or third
decade).
Note that immunocompetent adults (such as the
pregnant women described above) who are infected with
Toxoplasma gondii
often develop generalized lymph
node enlargement.
BIG PICTURE: In AIDS patients and fetuses
Toxoplasma gondii is
TOXIC
to the
BRAIN
and
EYES!!!
Diagnosis of toxoplasmosis can be made by:
1) CAT scan of brain will show a contrast-enhanc-
ing mass.
2) Examination of the retina of the eye will reveal
retinal inflammation.
3) Serology: Elevated immunoglobulin titers suggest
that the patient has at some time been exposed to this
organism.
Sulfadiazine plus pyrimethamine can be used to
treat patients with acute toxoplasmosis.
CHAPTER 30. PROTOZOA
23 9
Pneumocystis carinii
Pneumocystis carinii
is a flying-saucer appearing
FUNGUS that has previously been classified as a proto-
zoan but now has been shown to be more closely related
to fungi. This organism appears to invade the lungs of all
individuals at an early age and persists in a latent state.
In fact, based on IgM and IgG levels, it appears that
about 85% of children have had a mild or asymptomatic
respiratory illness with
Pneumocystis carinii
by age 4. In
persons with a functioning immune system, this organ-
ism will live comfortably within the lung without caus-
ing symptoms. However, in immunocompromised pa-
tients (AIDS patients, cancer patients, and organ trans-
plant recipients), this organism can multiply in the
lungs and cause a severe interstitial pneumonia, called
Pneumocystis carinii
pneumonia (PCP).
PCP is the most common opportunistic infection of
AIDS patients. Without prophylactic treatment there is
a 15% chance each year of infection, if the CD4+ T-
helper cell count is below 200. Clinically, this illness
presents with fever, shortness of breath, a nonproduc-
tive cough, and eventually death if not properly treated.
Chest X-ray may show diffuse bilateral interstitial in-
filtrates, or it can be normal.
The Case of the Breathless Woman Who Had
No Helpers
A 22 year-old female comes to the hospital with fever
and a feeling of chest tightness. She says she has no
medical problems but has lost weight. On physical ex-
amination you find large lymph nodes everywhere and
numerous genital warts. You note that she is tachy-
pneic, breathing 30 breaths per minute.
You look over her past record and find that she had a
child that was born with AIDS.
Her chest film shows diffuse perihilar interstitial
streaking bilaterally, sparing the outer lung margins.
You order an absolute T4-cell count and find that she
has 150 T-helper cells (Normal is greater than 1000).
Diagnosis of
Pneumocystis carinii
can be made by
silver-staining alveolar lung secretions, revealing the
flying saucer-appearing fungi. These secretions can be
obtained as follows, in order of increasing yield:
1) Induce a sputum sample by spraying saline into
the bronchioles and collecting the coughed material
(60% sensitivity).
2) Insert a fiberoptic camera (bronchoscope) deep
into the patient's bronchial tree, inject saline, and then
wash it out (bronchoalveolar lavage) (98% sensitivity).
3) Biopsy the lung by bronchoscopy (100% sensitivity).
About 80% of AIDS patients will get PCP at least
once in their lifetime unless prophylactic trimetho-
CHAPTER 30. PROTOZOA
Figure 30-6
prim/sulfamethoxazole is given when
CD4+
T-cell
counts drop below
200-250.
More than 60% of PCP in-
fections are being prevented with this prophylactic in-
tervention! Symptomatic patients can be treated with
high dose trimethoprim/sulfamethoxazole or intra-
venous pentamidine.
MALARIA
Plasmodium falciparum, Plasmodium vivax,
Plasmodium ovale,
and
Plasmodium malariae
Malaria is a febrile disease caused by
4
different pro-
tozoa:
Plasmodium falciparum, Plasmodium vivax, Plas-
modium ovale,
and
Plasmodium malariae.
They infect
about 300-500 million persons worldwide each year, re-
sulting in
20-40
million deaths. The anopheles mosquito
carries the organisms within its salivary glands and in-
jects them into humans while it feeds. The organisms
then grow in the liver and spread to the human red blood
cells, where they reproduce. The red cells fill with proto-
zoa and burst. The red cells all burst at the same time, re-
leasing the protozoa into the bloodstream and exposing
them to the immune system, which results in fever.
Fig. 30-6. The different species of
Plasmodium
burst the red cells at different time intervals.
Plus-
240
modium, vivax
and
Plasmodium ovale
burst loose
every
48
hours. The people who discovered all this
started numbering things with one (they didn't have a
zero) and so zero hours was called one,
24
hours
called two, and
48
hours called three. So, P.
vivax
and P.
ovale
produce chills and fever followed by
drenching sweats every
48
hours, which is called
tertian malaria. P.
malariae
bursts loose every 72
hours, causing a regular 3-day cycle of fevers and
chills, followed by sweats. This is called quartan
malaria. P.
falciparum,
the most common and deadly
of the
Plasmodia,
bursts red cells more irregularly,
between 36
48
hours. Thus the chills and fevers tend
to either fall within this period or be continuous, with
less pronounced chills and sweats.
Plasmodia Life Cycle
Fig. 30-7. The life cycle of the
Plasmodia
is complex,
since the protozoa divide into many different forms with
different names. This is clinically important because
the diagnosis of this disease rests on being able to iden-
tify these forms on a slide of a patient's blood.
Imagine yourself in Kenya with a patient who has in-
termittent shaking chills, fevers, and soaking sweats.
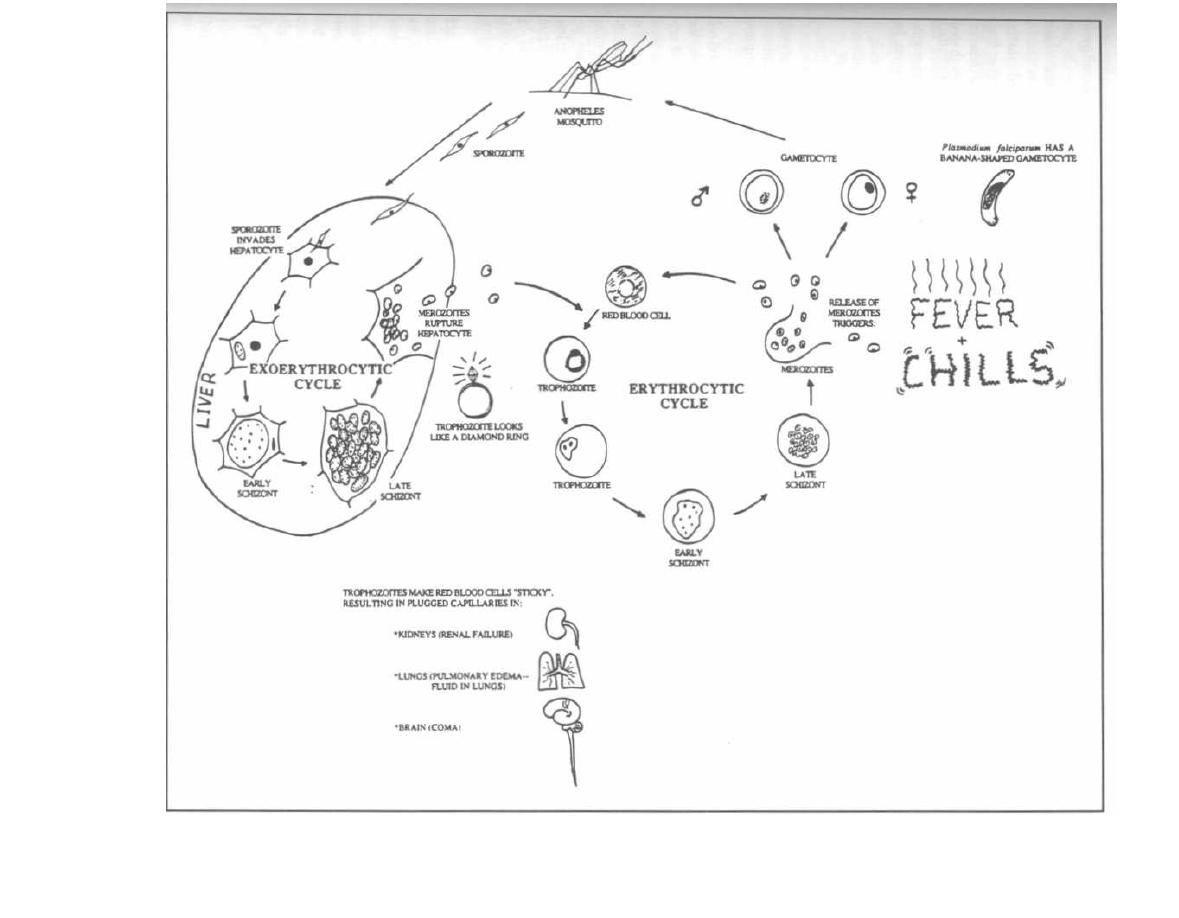
Figure 30-7

You pull out your little microscope with a reflecting mir-
ror light source, tilt it to catch some of the deep red
African sun, and focus your attention on the smear of
red cells in front of you. What are those life cycle stages
and what do they look like?
Plasmodia
undergo sexual division in the anopheles
mosquito and asexual division in the human liver and
red cells. Let's start with the human:
Thin, motile, spindle-shaped forms of the
Plasmodia,
called sporozoites, swim out of the mosquito's sucker
and into the human bloodstream. They wiggle their way
to the liver and burrow into a liver cell. This marks the
beginning of the pre-erythrocytic cycle in the liver,
so-named because this cycle occurs before the red blood
cells (erythrocytes) are invaded. The sporozoite rounds
up to form a ball within the liver cell. This ball, now
called a trophozoite, undergoes nuclear division, form-
ing thousands of new nuclei. This big mass is now a cell
with thousands of nuclei, called a schizont. A cytoplas-
mic membrane then forms around each nucleus, creat-
i ng thousands of small bodies called merozoites. The
new overloaded liver cell bursts open, releasing the
merozoites into the liver and bloodstream. Some will re-
i nfect other liver cells as the sporozoite did initially, re-
peating the same cycle discussed above, which is now
called the exo-erythrocytic cycle.
Other merozoites will enter the bloodstream and
enter red blood cells, starting the erythrocytic cycle.
This cycle is similar to the exo-erythrocytic cycle,
except that it occurs in the erythrocytes rather than
the liver cells. The merozoite rounds up to form a
trophozoite. In the red cells the trophozoite is shaped
like a ring with the nuclear material looking like the
diamond on the ring. Nuclear division then occurs with
formation of a large multinucleated schizont. Cyto-
plasm surrounds each nucleus to form new merozoites
within the late schizont. Red cell lysis occurs with
release of merozoites. The released merozoites stimu-
late an immune response, manifested as fevers, chills,
and sweats.
The merozoites can continue to invade other red cells
and then grow for another 2-3 day cycle followed by rup-
ture and release again. Some merozoites will change
into male and female gametocytes. These cells circu-
late and will be taken up by a biting anopheles mos-
quito. If they are not, they will shortly die.
Two of the species, P.
vivax
and P.
ovale,
produce dor-
mant forms in the liver (hypnozoites) which can grow
years later, causing relapsing malaria. This is why you
are asked if you have ever had malaria when you donate
blood (an effort to screen out infected blood).
In the mosquito, the gametocytes are sucked into the
stomach where the male and female gametocytes fuse.
DNA is mixed and the fused gametocytes become an
oocyst. The oocyst divides into many spindle-shaped
wiggling sporozoites, which disseminate within the
mosquito. They may find their way into the mosquito
salivary gland and will be injected into the human for
asexual reproduction.
The Disease Malaria
Malaria is well known for causing periodic episodes of
severe chills and high fevers along with profuse sweat-
ing at 48-72 hour intervals. These episodes commonly
last about 6 hours and are associated with the rupture
of red blood cells.
You can imagine that all these cycles of red blood cell
lysis must take their toll! In fact, P.
falciparum,
the
most aggressive species, will invade up to 30% of ery-
throcytes, which results in anemia and sticky red blood
cells. These sticky cells plug up post-capillary venules in
organs such as the kidney, lung, and even brain, result-
ing in hemorrhages and blocked blood delivery to those
tissues. Renal failure, lung edema, and coma may en-
sue, leading to death. Most deaths occur in children less
than 5 years old in sub-Saharan Africa. These children
often develop cerebral malaria characterized by
seizures and impaired consciousness, leading to coma.
Even with treatment, 20% of children with cerebral
malaria will die.
Infected individuals also get hepatomegaly and
splenomegaly. The spleen and liver enlarge as the fixed
phagocytic cells (of the reticuloendothelial system) pick
up large amounts of debris from the destroyed red blood
cells. The enlarged spleen occasionally ruptures.
Many African-American and African blacks are resis-
tant to P.
vivax
and
P. falciparum
infection. The resis-
tance to P.
vivax
is based on the absence of red cell
membrane antigens Duffy a and b that the P.
vivax
uses for binding. The sickle cell anemia trait (hemoglo-
bin AS) appears to help protect the red cells from P.
Figure 30-8
MALARIA
CHAPTER 30. PROTOZOA
242

I
falciparum
invasion. Endemic infection with malaria in
the African continent is thought to have led to a Dar-
winian selection process, resulting in high levels of
sickle trait and absence of Duffy a and b in many
African and African-American blacks.
Fig. 30-8.
Comparison of the
Plasmodia
species.
Diagnosis
1) Examination of thin and thick smears
(1000x)
of
blood, under oil-immersion magnification, reveals the
trophozoites and schizonts within the erythrocytes.
Sometimes the gametocytes can be visualized.
2) Fluorescently labeled antibodies may be used to
identify the responsible species.
Control of Malaria
1) Prevent mosquito bite:
a) Eliminate vector with pesticides (pyrethins) at
dusk in living and sleeping areas.
b) Use insect repellants (containing DEFT) and
mosquito nets, and wear long-sleeved shirts and
long pants.
2) Chemical Prophylaxis for travelers: When traveling
to an area without chloroquine resistance, chloroquine
is used. If in a chloroquine resistant area, mefloquine or
doxycycline may be used for prophylaxis.
It is wise to carry a pyrimethamine/sulfadoxine (fan-
sidar) "starter pack" to take in case of breakthrough in-
fection when far away from medical care. This is
especially true with doxycycline.
Fig. 30-9.
Chloroquine-resistant
P. falciparum
ar-
eas. Malaria is a disease of the tropics, cutting a swath
across the equator. Chloroquine-resistant
Plasmodium
falciparum
areas include most of Africa, Central Amer-
ica south of the Panama Canal, South America, India,
and South East Asia (see map). Chloroquine-sensi-
tive areas include North Africa, Central America North
of the Panama Canal, Haiti, and the Middle East.
Treatment of Malaria
To treat malaria you must understand two concepts:
1) the geographic pattern of susceptibility of P.
falci-
parum
to antimalarial drugs ( Fig.
30-9.)
and 2) the type
of
Plasmodium
species causing the infection.
1) P. malariae, P. vivax,
and P.
ovale
are all suscepti-
ble to chloroquine! Pushovers! But don't forget that P.
vivax and P.
ovale
have exo-erythrocytic cycles in the
liver and will be protected there from chloroquine. The
CHAPTER 30. PROTOZOA
243
Figure 30-9
acute infection will subside, but relapses will occur.
Treatment with
Primaquine
will kill the liver holdouts.
Primaquine is the Prime drug to kill
P. vivax
and P.
orale
in the liver!
2) Plasmodium falciparum:
This guy is nasty, caus-
ing the most hemolysis, organ damage, and death.
a) Chloroquine-sensitive areas: Huh, I wonder
which drug to use?? Chloroquine alone is enough
as P.
falciparum
does not have an exo-erythrocytic
cycle.
b) Chloroquine-resistant areas: Treatment options
include quinine (quinidine, the antiarrhythmic
drug, is more expensive but readily available in the
United States and just as effective), artemether
(see below), pyrimethamine/sulfadoxine, or
mefloquine. Severe infection (cerebral malaria) is
treated with IV or IM quinine, quinidine, or
artemether.
Newsflash!!! Artemether is new therapy for se-
vere falciparum malaria in children and adults!
Chloroquine-resistant P.
falciparum
causes severe
malaria in Africa, killing about 500,000 children a year
(1-2 million world-wide). Quinine is the preferred ther-
apy in these areas because it can be injected intramus-
culary (IM). A new drug named artemether (or its
brother artesunate) is derived from a traditional Chi-
nese malaria remedy (qinghaosu or wormwood!).
Artemether is effective against chloroquine-resistant P.
falciparum
and has proven to work as well as quinine in
the treatment of severe malaria. Unfortunately, even
with therapy, 20% of children with cerebral malaria still
die. (Hoffman, 1996; Van Hensroek, 1996; Hien, 1996;
White, 1996).
There are a number of common features of these
drugs (also see Fig.
30-13).
1) All of the anti-malarial drugs can be taken orally.
2) All of the anti-malarial drugs cause GI upset as a
primary adverse effect.
3) Chloroquine, primaquine, and quinine all cause
hemolysis in patients with glucose-6-phosphate dehy-
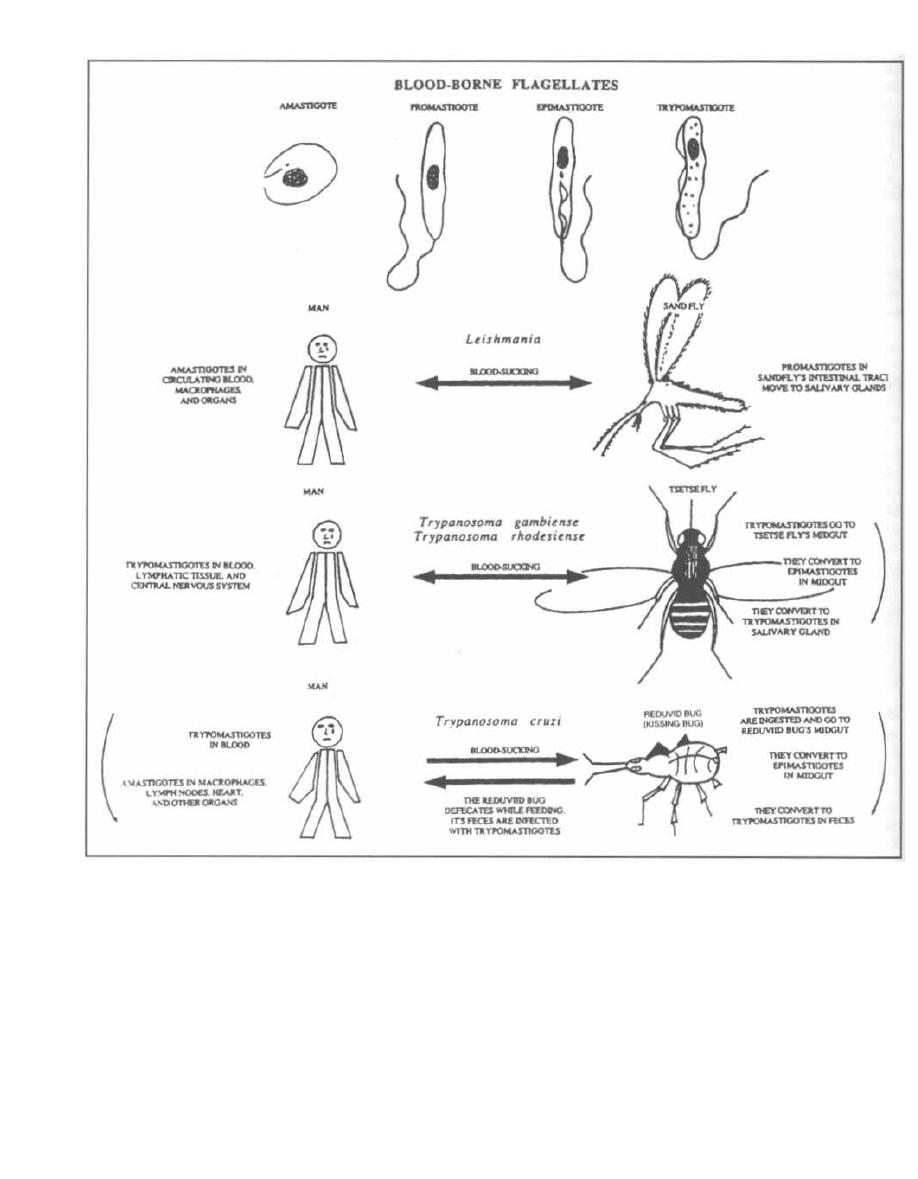
Figure 30-10
drogenase deficiency (G-6-P-D deficiency is present
in some Africans, Mediterraneans, and Southeast
Asians).
4) Chloroquine, quinine, quinidine, and sulfadoxine/
pyrimethamine are safe in all trimesters of
pregnancy. Not enough data is available about the
others.
CHAPTER 30. PROTOZOA
24 4
BABESIOSIS
( Babesia microti, Babesia divergens )
Babesiosis is an infection very much like malaria. It is
transmitted by the bite of a blood sucker (tick in this case)
and it invades and can be seen inside, red blood cells. It
also causes fever and hemolysis (anemia), as in malaria.

It is different in that:
1) There are more than 100 species of Babesia, mostly
causing disease in cattle and other domestic or wild
animals.
2) Babesia are spread by tick bites, not mosquito bites.
3) They do not affect liver cells (so there is no exoery-
throcytic phase).
In the northeastern coastal United States (e.g. Nan-
tucket Island) Babesia Microti is spread by the bite of
the same tick that spreads lyme disease, Ixodes scapu-
laris. After biting the white-footed mouse, the reservoir
for B. microti, the tick will leap to the next carefree
golfer who walks into the rough.
Like Plasmodium, Babesia sporozoites slither out of
tick salivary glands into the blood of the hapless golfer.
The sporozoites invade erythrocytes and differentiate
into pear or ring-shaped trophozoites. Trophozoites
asexually bud and divide into 4 merozoites that stick to-
gether, forming a cross or x-shaped tetrad ("Maltese
cross"). Red cell infection results in only mild hemolysis,
so infection is usually asymptomatic and sub-clinical.
Asplenic patients are unable to clear the organisms as
well and may have severe infection similar to falci-
parum malaria. (Gelfand, 1995; Persing, 1995). Treat
infected patients with quinine and clindamycin.
Fig. 30-9A. Babesia sporozoites in the "hood".
Giemsa or wright-stained thin and thick blood smears
reveal ring-shaped trophozoites that look like Plasmod-
ium and the distinctive cross or x-shaped tetrad of
merozoites (Maltese cross). Transmitted by tick vector.
THE BLOOD-BORNE FLAGELLATES
(Leishmania and Trypanosoma)
Fig. 30-10. Leishmania and Trypanosoma are trans-
mitted by the bite ofa blood-sucking insect. Although they
CHAPTER 30. PROTOZOA
245
cause different diseases, they pass through similar mor-
phologic states in the human and insect host. They can ex-
ist as rounded cells without flagella, called amastigotes,
or as flagellated motile forms called promastigotes, epi-
mastigotes, and trypomastigotes. These are named
according to the insertion site oftheir single flagellum. All
ofthese organisms cause an initial skin ulcer at the site of
the insect bite, followed by systemic invasion.
These parasites can also be transmitted via blood
product transfusion.
Leishmaniasis
(Leishmania tropica, Leishmania chagasi, Leishmania
major, Leishmania braziliensis, Leishmania donovani)
Leishmania is zoonotic, carried by rodents, dogs, and
foxes, and is transmitted to humans by the bite of the
sandfly. The disease leishmaniasis is found in South
and Central America, Africa, and the Middle East.
Following transmission from the sandfly, the pro-
mastigote invades phagocytic cells (macrophages) and
transforms into the nonmotile amastigote. The amastig-
ote multiplies within the phagocytic cells in the lymph
nodes, spleen, liver, and bone marrow (the reticuloen-
dothelial system) (see Fig. 30-10).
The different diseases caused by Leishmania depend
on the invasiveness of the species as well as the host's
immune response. Host immunity depends on a cell-me-
diated defense. It appears that some patients have ge-
netically deficient defenses against Leishmania and
will be afflicted with more severe disease. Leishmania-
sis presents in a spectrum of disease severity: from a
single ulcer that will heal without treatment; to widely
disseminated ulcerations of the skin and mucous mem-
branes; to the very severe infection striking deep into
the reticuloendothelial organs, the spleen and liver.
Note the similarity here to leprosy (see Chapter 14) in
which differences in host cell-mediated defenses result
in varied severity of disease.
There are 3 clinical forms of this disease:
1) Cutaneous leishmaniasis
a) Simple cutaneous lesions
b) Diffuse cutaneous lesions
2) Mucocutaneous leishmaniasis
3) Visceral leishmaniasis
Cutaneous Leishmaniasis
A sandfly injects Leishmania into the skin, where
they migrate to reticuloendothelial cells (fixed phago-
cytic cells in lymph nodes). At the site of the sandfly
bite, a skin ulcer develops, called an "oriental sore." This
ulcer heals in about a year, leaving a depigmented (pale)
scar. Diagnosis is made by observing Leishmania in
stained skin-scrapings from the ulcer base.
Cell-mediated immunity is intact, so the immune sys-
tem attacks the organisms resulting in skin destruction
(ulcer formation) and clearance of infection (similar to
the situation with tuberculoid leprosy). Because of the
intact cell-mediated immunity, this organism invokes a
delayed hypersensitivity reaction. Diagnosis can be
made by injecting killed
Leishmania
intradermally
(Leishmania skin test). Just like the PPD test of tuber-
culosis, a raised indurated papule 48 hours later sup-
ports the presence of a
Leishmania
infection.
This disease is also seen in Latin America and Texas,
where it is called American cutaneous leishmaniasis.
Diffuse Cutaneous Leishmaniasis
In Venezuela and Ethiopia, a chronic form of cuta-
neous leishmaniasis occurs in patients with deficient
immune systems. A nodular skin lesion arises but does
not ulcerate. With time, numerous nodular lesions arise
diffusely across the body. There is often a concentration
of lesions near the nose. The untreated infection can last
more than 20 years.
The disease is diffuse because the host's immune
system does not respond to the invasion by
Leishma-
nia,
due to a defect in cell-mediated immunity. There-
fore, the promastigotes are able to spread and infect
the skin, causing the diffuse nodular lesions. Due to
the defect in cell-mediated immunity, the Leishmania
skin test is negative (similar to the situation in lepro-
matous leprosy).
Mucocutaneous Leishmaniasis
Initially, a dermal ulcer, similar to cutaneous leishma-
niasis, arises at the site of the sandfly bite and soon heals.
However, months to years later, ulcers in the mucous
membranes of the nose and mouth arise. If untreated, the
infection is chronic, with erosion of the nasal septum, soft
palate, and lips, over a course of 20-40 years. Death by
bacterial secondary infection may occur.
Diagnosis is made via skin scrapings.
Visceral Leishmaniasis (Kala-azar)
The sandfly transmits
Leishmania donovani
or
Leish-
mania chagasi
to an individual (most commonly young
malnourished children), who months later will complain
of abdominal discomfort and distension, low-grade
fevers, anorexia, and weight loss. This abdominal en-
largement is due to
Leishmania donovani's
invasion of
the reticuloendothelial cells (fixed phagocytic cells) of the
spleen and liver, causing hepatomegaly and massive
splenomegaly. Patients also develop a severe anemia
and the white blood-cell count can also be very low. Most
cases (over 90%) are fatal if untreated.
Diagnosis is made by liver and spleen biopsies
demonstrating these protozoa. The Leishmania skin
CHAPTER 30. PROTOZOA
246
test is negative during active disease as cell-mediated
immunity is deficient.
All forms of leishmaniasis can be treated with the
pentavalent antimonial stibogluconate.
African Sleeping Sickness
(Trypanosoma brucei rhodesiense
and
Trypanosoma
brucei gambiense)
Trypanosoma rhodesiense
and
Trypanosoma gambi-
ense
are responsible for African sleeping sickness, which
is transmitted by the blood-sucking bite of a tsetse fly.
Following this bite, the motile flagellated form of these
2 organisms, called a trypomastigote, spreads via the
person's bloodstream to the lymph nodes and central
nervous system (CNS) (see Fig. 30-10).
The first manifestation is a hard, red, painful skin ul-
cer that heals within 2 weeks. With systemic spread, the
patient then experiences fever, headache, dizziness, and
lymph node swelling. These symptoms can last a week,
and then the fever subsides for a few weeks followed by
renewed fevers. This pattern of fevers with fever-free in-
tervals can occur for months. Finally, CNS symptoms
develop, with drowsiness in the daytime (thus sleeping
sickness), behavioral changes, difficulty with walking,
slurred speech, and finally coma and death.
There are 2 forms of African sleeping sickness. West
African sleeping sickness, caused by
Trypanosoma
brucei
gambiense, is notable for slowly progressing
fevers, wasting, and late neurologic symptoms. East
African sleeping sickness, caused by
Trypanosoma
brucei rhodesiense,
is similar to the West African vari-
ety but more severe, with death occurring within weeks
to months. There is rapid progression from recurrent
fevers to early neurologic disease (drowsiness, mental
deterioration, coma, and death).
Q: Why the intermittent fevers???
A: Variable surface glycoproteins (VSG). The
trypanosomes are covered with about 10 million mole-
cules of a repeating single glycoprotein called the VSG.
The trypanosomes possess genes that can make thou-
sands of different VSGs. They will make and express, on
their surface, a new VSG in a cyclical nature. Every
time the human host develops antibodies directed
against the VSG (and fever with this immune recogni-
tion), the trypanosomes produce progeny with a new
VSG coat. Thus, there are waves of new antigens, pro-
ducing recurrent fevers and protection from our im-
mune defenses. This is similar to the antigenic variation
of the spirochete
Borrelia recurrentis,
which causes re-
lapsing fever (see Fig. 13-12).
Diagnosis consists of visualization of trypomastigotes
in peripheral blood, lymph nodes, or spinal fluid.

Figure 30-11
Patients are treated with suramin if the central ner-
vous system (CNS) is not involved (suramin does not
penetrate into the CNS). With CNS involvement, the ar-
senical melarsoprol, which is extremely toxic, is used.
Chagas' Disease
(Trypanosoma cruzi ,
the American Trypanosome)
Chagas' disease is caused by a trypanosome, but the
pathogenesis and epidemiology differ greatly from the
African trypanosomes.
This is truly a disease of the Americas, ranging from the
southern U. S. (Texas), Mexico, Central America, and
down into South America.
T. cruzi
survives in wild animal
reservoirs such as rodents, opossums, and armadillos.
The vector is the reduviid bug, also called the kissing
bug. The bug feeds on humans while they sleep and defe-
CHAPTER 30. PROTOZOA
24 7
Cates while it eats.
T. cruzi
trypo-mastigotes, which are
present in the bug's feces, tunnel into the human host.
The trypomastigote loses its undulating membrane and
flagellum and rounds up to form the amastigote, which
rapidly multiplies. Organisms invade the local skin,
macrophages, lymph nodes, and spread in the blood to dis-
tant organs (see Fig. 30-10).
Acute Chagas' Disease
At the skin site of parasite entry, a hardened, red
area develops, called a chagoma. This is followed by
systemic spread with fever, malaise, and swollen
lymph nodes. Organs that can be infected include the
heart and central nervous system (CNS). Heart in-
flammation results in tachycardia and electrocardio-
graphic changes, while the CNS involvement can

result in a severe meningoencephalitis (usually in
young patients).
This acute illness resolves in about a month and pa-
tients then enter the intermediate phase. In this
phase there are no symptoms, but there are persistently
low levels of parasite in the blood as well as antibodies
against T. cruzi. Most persons will remain in the inter-
mediate phase for life.
For reasons that are poorly understood, some per-
sons will develop chronic Chagas' disease years to
decades later.
Chronic Chagas' Disease
The organs primarily affected are the heart and some
hollow organs such as the colon and esophagus. Intra-
cellular T. cruzi amastigotes cannot usually be found,
and it is unclear why disease develops in these organs.
1) Heart: Arrhythmias are the earliest manifesta-
tion (heart block and ventricular tachycardia). Later
there is an increase in heart size and heart failure (di-
lated cardiomyopathy).
2) Megadisease of colon and esophagus: A big,
dilated, poorly functioning esophagus develops with
symptoms of difficulty and pain in swallowing, and re-
gurgitation of food. A dilated colon (megacolon) results
in constipation and abdominal pain. Patients can go
weeks without bowel movements.
Fig. 30-11. Trypanosoma cruzi (the American Try-
panosome). To remember that T. cruzi causes mega-
colon, electrical arrhythmias ,and dilatation of the
heart, and is transmitted by the feces of the kissing bug,
picture Tom Cruise (the American actor).
Diagnosis and Treatment
Acute Chagas' disease:
1) Direct examination of the blood for the motile try-
pomastigotes.
2) Xenodiagnosis: This sensitive test is conducted
as follows. Forty laboratory-grown reduviid bugs are al-
lowed to feed on the patient, and one month later the
bugs' intestinal contents are examined for the parasite.
Chronic Chagas' disease:
Classic clinical findings (cardiac and megadisease)
along with serologic evidence of past T. cruzi infection
allows for presumptive diagnosis.
Although nifurtimox and benznidazole can be used
for acute cases, there is currently no effective therapy for
the chronic manifestations of this infection. Therefore,
CHAPTER 30. PROTOZOA
248
individuals should take precautions to prevent kisses by
the kissing bug (insect repellent, bednets).
Balantidium Coli
If you do not want diarrhea, do not consume food or
water contaminated by pig feces! This advice will pre-
vent ingestion of
B.
coli cysts. These cysts mature into
ciliated trophozoites, and travel to the intestinal tract.
The trophozoites dig into the intestinal wall, where they
exist happily consuming the native bacteria. Most in-
fected individuals are asymptomatic, while some will
develop diarrhea.
B. Coli
trophozoites are notable for being the largest
parasitic protozoans found in the intestine. Diagnosis is
made by identifying the ciliated trophozoites or cysts in
stool specimens. Tetracycline is effective at treating this
infection.
Fig. 30-12.
Summary of the protozoan diseases.
Fig. 30-13. Treatment of protozoan diseases.
References
Dupont HL, Chappell CL, et al. The infectivity of
Cryp-
tosporidium paruum in healthy volunteers. N Engl J Med
1995;332:855-9.
Gelfand JA. Babesia. In: Mandell GL, Bennett JE, Dolin R,
eds. Principles and Practice of Infectious Disease. 4th ed.
New York: Churchill Livingstone, 1995:2415-2427.
Hein TT, Day NP, et al. A controlled trial of artemether or qui-
nine in Vietnamese adults with severe falciparum malaria.
N Engl J Med 1996;335:76-83.
Hoffman SL. Artemether in severe malaria-still too many
deaths. N Engl J Med 1996;335:124-5.
Kirchhoff LV. American trypanosomiasis (Chagas' disease) -a
tropical disease now in the United States. N Engl J Med
1993;329:639-644.
Krogstad DJ. Plasmodium species (malaria). In: Mandell GL,
Bennett JE, Dolin R, eds. Principles and Practice of Infec-
tious Disease. 4th ed. New York: Churchill Livingstone,
1995:2415-2427.
Persing DH, Herwaldt BL, et al. Infection with a Babesia-like
organism in northern California. N Engl J Med 1995;
332:298-303.
Rosenblatt JE. Antiparasitic Agents. Symposium on Antimi-
crobial Agents-Part VII. Mayo Clin Proc 67:276-287, 1992.
Sanford JP, Gilbert DN, et al. Guide to Antimicrobial Therapy
1996. Antimicrobial Therapy, Inc, Dallas Texas; 1996.
Ungar BL. Cryptosporidium. In: Mandell GL, Bennett JE,
Dolin R, eds. Principles and Practice of Infectious Disease.
4th ed. New York: Churchill Livingstone, 1995:2500-2510.
Van Hensbroek MB, Onyiorah E, et al. A trial of artemether
or quinine in children with cerebral malaria. N Engl J Med
1996;335:69-75.
White NJ. Current concepts: The treatment of malaria. N
Engl J Med 1996;335:800-06.
Wyler DJ. Malaria chemoprophylaxis for the traveler. N Engl
J Med 1993;329:31-37.
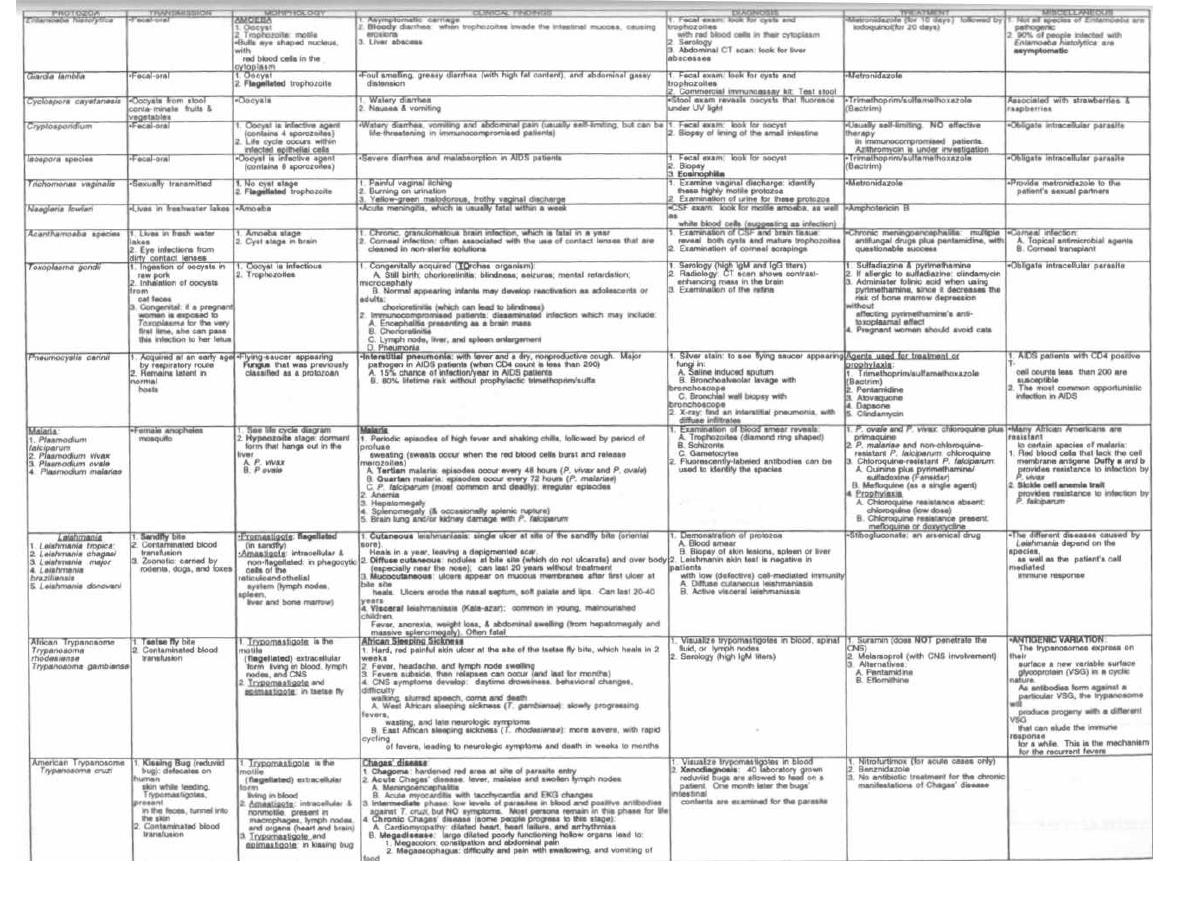
Figure 30-12
PROTOZOA
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple ®MedMaster
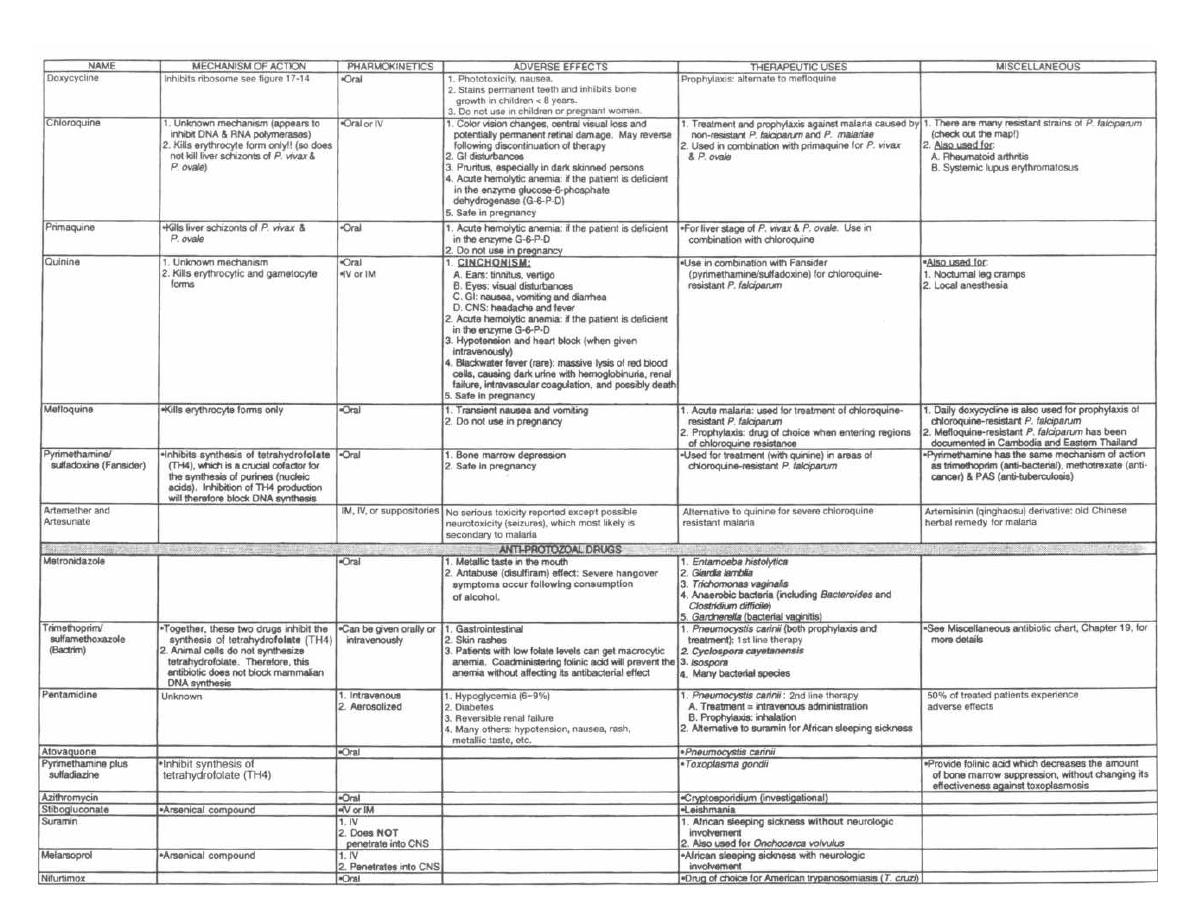
Figure 30-13
ANTI-PARASITIC DRUGS
M. Gladwin and B. Trattler, Clinical Microbiology Made Ridiculously Simple OMedMaster

CHAPTER 31.
Helminths is Greek for "worm." Worms are usually
macroscopic, although diagnosis often requires the vi-
sualization of the eggs, which are microscopic, in the
stool. We will discuss 16 types of worms that cause sig-
nificant infections in humans. The first 10 are round-
worms (nematodes), and the last 6 are the more
primitive flatworms (platyhelminthes). By under-
standing their life cycles, you can learn ways to prevent
or eradicate helminthic infections.
Within the normal human host there is usually
no immune reaction to living worms. However, there
is often a marked response to dead worms or eggs. With
many of the worm infections, our immune system is
kind enough to raise a red flag-elevating the level of
eosinophils in the blood-thereby assisting in diagnosis.
NEMATODES
(Roundworms)
Intestinal Nematodes
"Intestinal" nematodes all mature into adults within
the human intestinal tract. The larval forms of many of
these roundworms may be distributed widely through-
out the body.
Three of the intestinal nematodes are acquired by the
i ngestion of eggs: Ascaris lumbricoides, Trichuris
trichiura ( whipworm), and Enterobius vermicularis
( pinworm). Two worms, Necator americanus ( hook-
worm) and Strongyloides stercoralis, are acquired when
their larvae penetrate though the skin, usually of the
foot. Trichinella spiralis is acquired by the ingestion of
encysted larvae in muscle (pork meat).
With infection, most of these intestinal worms (except
for Enterobius and Trichuris, which stay in the intesti-
nal tract) invade other tissues at some stage of their life
cycle. This stimulates our immune system to raise the
number of eosinophils in the blood.
Fig. 31-1.
The first 3 roundworms (Ascaris lumbri-
coides, Necator americanus, and Strongyloides sterco-
rous)
all have a larval form that migrates through
the tissue and into the lung at some stage of their life
cycle. The larvae grow in the lung, are coughed up and
swallowed into the intestine, where they grow into
adult worms.
1) Ascaris lumbricoides:
Infection occurs in the
tropics and mountainous areas of the southern U. S.,
when individuals consume food that is contaminated
with eggs. Larvae emerge when the eggs reach the
small intestine. The larvae penetrate through the in-
CHAPTER 31. HELMINTHS
251
HELMINTHS
testinal wall and travel in the bloodstream to the lungs.
The larvae grow in lung alveoli until they are coughed
up and swallowed. These larvae again reach the small
i ntestine and mature into adults. Here each adult worm
produces over 200 thousand eggs per day, which are ex-
creted in feces.
2)
Necator americanus:
The larval form lives
in the soil and eats bacteria and vegetation. After
a week it transforms into a long, slender filari-
form larva that can penetrate human skin. The
filariform larva penetrates between the toes of the
hapless human who walks by shoeless. The larvae travel
directly to the alveoli of the lungs, where they grow and
are coughed up and swallowed. The adult worms de-
velop as they arrive at the small intestine, where they
attach by their mouths, and suck blood. At this point,
the hookworms copulate and release fertilized eggs.
3)
Strongyloides stercoralis:
The larval forms in
the soil penetrate the human skin and travel to the lung.
There they grow, are coughed up and swallowed into the
small intestine, where they develop into adult worms
that lay eggs. The eggs are not passed in the stools.
Rather, filariform larvae hatch and can do 3 things:
a) Autoinfection: The filariform larvae pene-
trate the intestine directly, without leaving, and go
to the lung to continue the cycle.
b) Direct cycle: The filariform larvae pass out in
the feces, survive in the soil, penetrate the next
passerby, and migrate to the lungs. This complete
cycle is almost exactly the same as that
of
Necator
americanus ( hookworm).
c) Indirect cycle: This is a sexual cycle where
the filariform larvae are passed out in the stool and
while in the soil develop into male and female
adults. They mate in the soil and produce fertilized
eggs. The filariform larvae then hatch and reinfect a
human, moving to the lung.
Ascaris lumbricoides
Fig. 31-1.
Ascaris infection may be mild or asympto-
matic. With heavy infections the patient may develop ab-
dominal cramping. Severe infections involve adult worm
invasion into the bile ducts, gall bladder, appendix, and
liver. Children with heavy worm loads may suffer from
malnutrition because the worms compete for the same food
and sometimes a mass of worms can actually block the in-
testine. When the larvae migrate into the lung, the patient
may develop cough, pulmonary infiltrate on chest
x-ray, and a high eosinophil count in the blood and sputum.
Diagnosis is made by identification of eggs in feces. A
sputum examination may reveal larvae. The peripheral
blood smear may also reveal an increased number of
eosinophils.
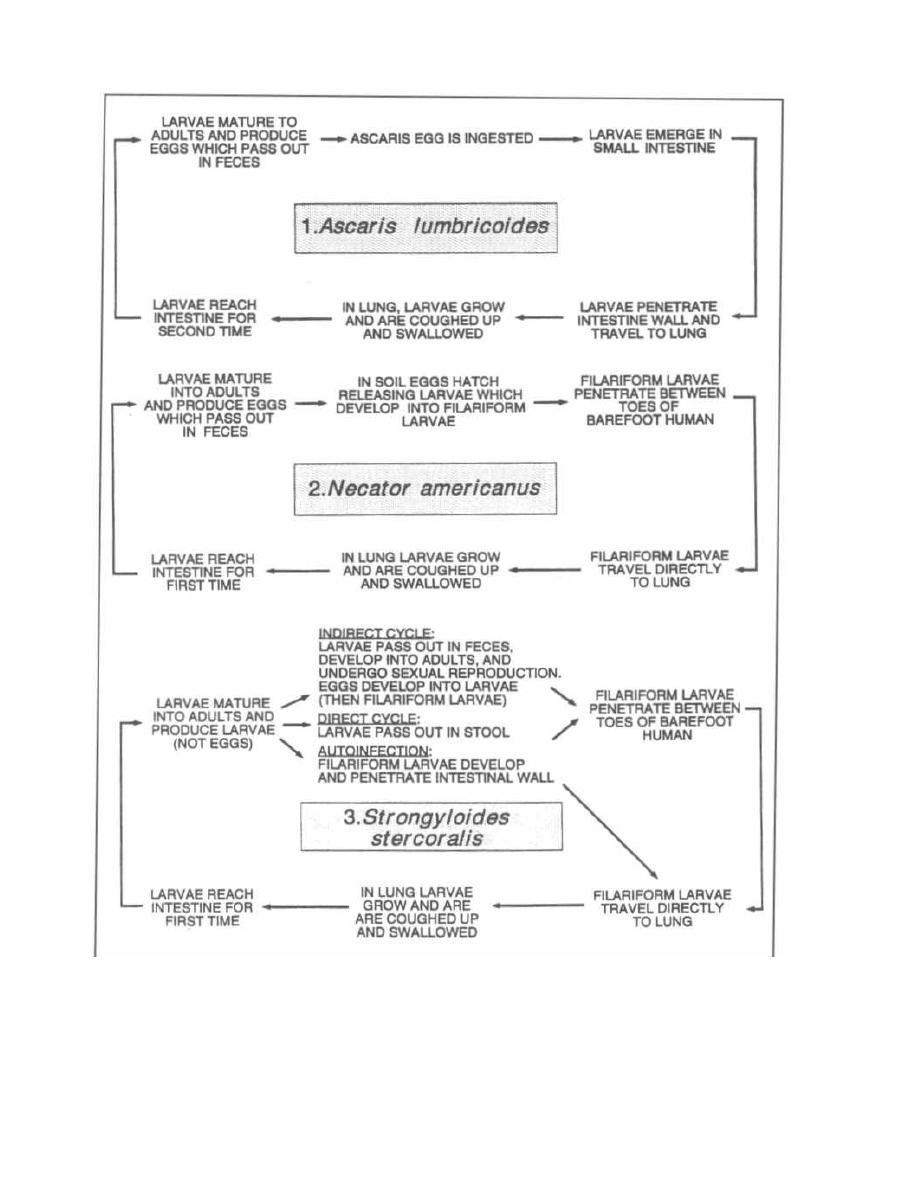
Figure 31-1
Treatment
The intestinal nematodes Ascaris lumbricoides,
Necator Americanus (hookworm), Strongyloides sterco-
CHAPTER 31. HELMINTHS
252
ralis, Enterobius vermicularis (pinworm), Trichuris
trichiura (whipworm), and Trichinella spiralis are all
treated with the same drug:

Think of a worm,
Worms BEND,
BEND them a lot and you can kill them!
Mebendazole
Thiabendazole
Albendazole
These drugs paralyze the roundworms so they are
passed out in the stool. Other drugs will irritate the
worms, causing them to migrate out of the small intes-
tine to other organs, which can be fatal.
Pyrantel pamoate is an alternative agent for En-
terobius (pinworm), Necator (hookworm), and Ascaris.
Necator americanus and Ancyclostoma
duodenale
(Hookworm)
Fig. 31-1. Notice that Ascaris lumbricoides and Neca-
tor americanus (hookworm) have very similar life cy-
cles. They differ only in the path that each larvae form
takes to reach the lung: Necator, foot to lung; Ascaris,
intestine to lung.
The patient with hookworm can develop diarrhea, ab-
dominal pain, and weight loss. Since each hookworm
sucks blood from the wall of the intestine, hookworm in-
fection may cause an iron deficiency anemia. There is
also an intense itching and rash at the site of penetra-
tion through the skin (between the toes), and the local
growth in the lung can result in a cough, infiltrate on
chest x-ray, and eosinophilia.
Diagnosis is made by identifying eggs in a fresh fecal
sample. This infection may be treated with mebenda-
zole. Also treat the iron deficiency anemia.
Strongyloides stercoralis
Fig. 31-1. Individuals infected with Strongyloides
may complain of vomiting, abdominal bloating, diarrhea,
anemia, and weight loss. Similar to hookworm infection,
patients may develop a prurititic rash, lung symptoms
(cough or wheezing), and/or eosinophilia. When patients
infected with strongyloides are given immunosuppres-
sive medications, such as prednisone, they can develop
a severe autoinfection. The filariform larvae will invade
the intestine, lung and other organs, causing pneumonia,
ARDS, and multi-organ failure. Patients with COPD
and asthma living in areas endemic for Strongyloides
should have their stool and eosinophil count checked be-
fore steroid treatment!
Diagnosis is made by identifying larvae in feces (no
eggs). The enterotest, where a long nylon string is
swallowed and later pulled back out the mouth, may
demonstrate larvae of
Strongyloides.
Sputum exam
CHAPTER 31. HELMINTHS
253
may also demonstrate larvae. Examination of the blood
will reveal an elevated level of eosinophils.
Treat infected patients with thiabendazole or al-
bendazole and ivermectin
Trichinella spiralis
You may have seen Gary Larson's "The Far Side" comic
showing wolves at the edge of a pork farm, saying, "Let's
rush them and Trichinella be damned." After reading
about Trichinella spiralis, we can finally get the joke.
Infection occurs following the ingestion of the encysted
larvae of Trichinella spiralis, which are often present in
raw pork. After ingestion, the cysts travel to the small in-
testine, where the larvae leave the cysts and mature into
mating adults. Following mating, the adult males are
passed in the feces. The females penetrate into the in-
testinal mucosa, producing thousands of larvae. The lar-
vae then enter the bloodstream and spread to organs and
skeletal muscle. The larvae then become encysted in
skeletal muscle, where they may last for decades.
Most patients are asymptomatic with the initial
infection. Some patients will complain of abdominal
pain, diarrhea, and fever as the worms mature in the
small intestine and penetrate through the intestinal
wall. About 1 week after the initial intestinal invasion,
the larvae migrate into skeletal muscle, producing
fevers and muscular aches. In severe (sometimes fatal)
cases, larvae may invade heart muscle and brain tissue.
Diagnosis can be made with serologic tests or muscle
biopsy, which will reveal the encysted larvae. Since there
is significant muscle invasion, examination of the differ-
ential white blood cell count will reveal a marked increase
in the percentage of eosinophils. The invasion of muscle
also results in increased levels of serum muscle enzymes,
such as creatinine phosphokinase (CPK).
Prevention is best carried out by killing cysts, either by
cooking or freezing the pork meat prior to consumption.
Mebendazole and thiabendazole have little effect
on muscle larvae but may be helpful.
Trichuris trichiura
(Whipworm)
The next 2 worms, Trichuris trichiura and Enterobius
vermicularis, have very simple life cycles: there is no fi-
lariform larvae stage, no tissue invasion, and no lung in-
volvement. Since there is no tissue invasion, the
eosinophil count is not elevated.
Fig. 31-2.
Trichuris trichiura has a simple slow
life cycle. Like the Congressional whip trying to move
his party to a decision, the whipworm life cycle is
slow. Transmission occurs by ingestion of food conta-
minated with infective eggs. The eggs hatch in the
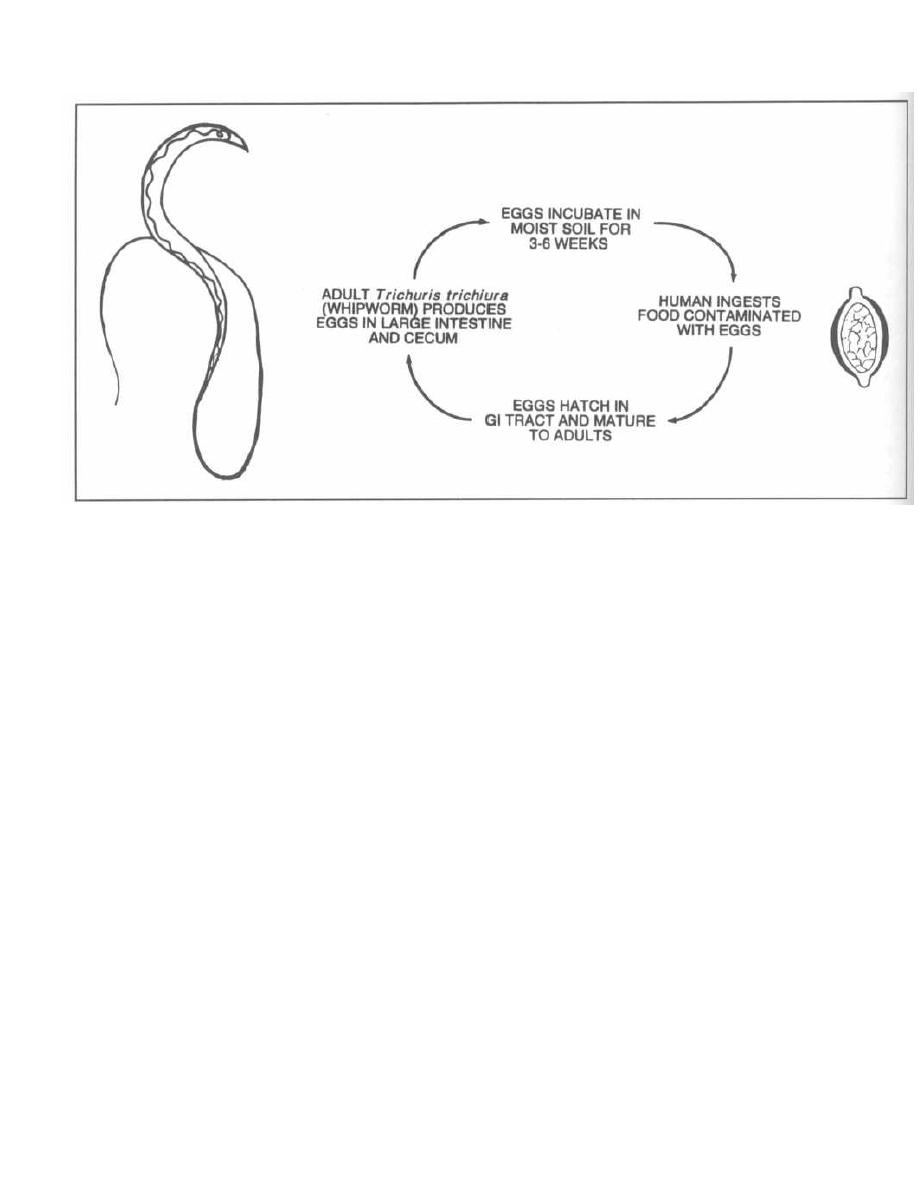
Figure 31-2
gastrointestinal tract and migrate to the cecum and
ascending large intestine. The mature adult will then
produce thousands of eggs per day for about one year.
There is no autoinfection, since the eggs must incu-
bate in moist soil for 3-6 weeks before they become
infective.
Infected patients may have abdominal pain and diar-
rhea. Diagnosis is made by identifying eggs in fecal
specimens. The eggs look like footballs with bumps on
each end. The adults have the appearance of a bullwhip,
thus the common name.
Treat patients with mebendazole.
Enterobius vermicularis
( Pinworm)
The
Enterobius'
life cycle is very simple: The eggs are
simply ingested, and the pinworms mature in the cecum
and ascending large intestine. The female migrates to
the perianal area (usually at night) to lay her eggs,
which become infectious 4-6 hours later. This infection
causes severe perianal itching. An infected individual
will scratch the perianal area and then reinfect himself
or others (hand-to-mouth) because his hands are now
covered with the microscopic pinworm eggs.
This infection is more of a nuisance than it is danger-
ous. The infected individual has a terribly itchy behind,
usually at night.
Diagnosis is made by placing scotch tape firmly on the
perianal area. The scotch tape will pick up eggs, which
CHAPTER 31. HELMINTHS
25 4
can be viewed under a microscope. At night the larger
adult females can sometimes be seen with the unaided
eye, crawling across the perineal area. There is no
eosinophilia, since there is no tissue invasion.
Treat with mebendazole or pyrantel pamoate,
avoid scratching, and change the sheets daily.
Fig. 31-3. Enterobius v
ermicularis . The female worm is
like an intestinal (entero) bus. She drives out of the
anus every night and drops off her egg passengers on
the perineum. If a pin (pinworm) pops her tire, seal
the leak with some scotch tape. The scotch tape test is
used to diagnose
Enterobius vermicularis
i nfection.
Blood and Tissue Nematodes
The blood and tissue nematodes are not spread by fe-
cal contamination, but rather by the bite of an arthro-
pod. These threadlike, round worms belong to the
family Filarioidea and so are called filariae. Adult fi-
lariae live in the lymphatic tissue and give birth to
prelarval forms (they do not lay eggs) called microfi-
lariae. The microfilariae burrow through tissue and
circulate in the blood and lymphatic system. Microfilar-
iae are picked up by bloodsucking arthropods, which
transmit them to another human. Disease is caused
by allergic reactions to both the microfilariae and
dead adult worms in the lymphatic system as well as
other organs (such as the eyes with
Onchocerca volvu-
lus
i nfection).

Figure 31-3
Onchocerca volvulus
This filarial infection is found in Africa and Central
and South America. The larvae are transmitted to hu-
mans by the bite of infected black flies. The microfi-
lariae (larvae) mature into adults, which can be found
coiled up in fibrous nodules in the skin and subcuta-
neous tissue. After mating, the adults produce micro-
filariae, which migrate through the dermis and connec-
tive tissue. Patients will develop a pruritic skin rash
with darkened pigmentation. The skin may thicken
with the formation of papular lesions that are actually
intraepithelial granulomas. The thickened skin may
appear dry, scaly, and thick ("lizard skin"). Microfilar-
iae may also migrate to the eye, causing blindness
(since the black fly vector breeds in rivers and streams,
this is often referred to as "river blindness"). There
are villages in endemic areas where most of the inhabi-
tants are blind.
Diagnosis is made by demonstrating microfilariae in
superficial skin biopsies, or adult worms in a nodule.
Microfilariae can often be seen in the eye (cornea and
anterior chamber) by slit lamp examination.
A new drug, ivermectin, has revolutionized the
treatment of Onchocerciasis. It kills microfilariae and
prevents the microfilariae from leaving the uteri of
adult worms. The manufacturer (Merck) has donated
the drug to the World Health Organization for a pro-
gram to eradicate Onchocerca from the planet. As hu-
CHAPTER 31. HELMINTHS
255
mans are the only reservoir, treating people in endemic
areas for 10 years (as planned) will prevent the birth of
new microfilariae while all the adult worms (which have
long life spans) die of old age.
If you know Spanish, ver means "to see." So: I VER
with IVERmectin!!
Wuchereria bancrofti
and
Brugia malayi
( Elephantiasis)
Wuchereria bancrofti and Brugia malayi both cause a
lymphatic infection that can result in chronic leg
swelling. Wuchereria infection is endemic to the Pacific
Islands and much of Africa, while Brugia is endemic to
the Malay Peninsula and is also seen in much of South-
east Asia. Transmission occurs by the bite of an infected
mosquito. The transmitted microfilariae mature into
adults within the lymphatic vessels and lymph nodes of
the genitals and lower extremities. Mature adults mate,
and their offspring (microfilariae) enter nearby blood
vessels.
Small infections may only result in enlarged lymph
nodes. Frequent infections in endemic areas result in
acute febrile episodes, associated with headaches and
swollen inguinal lymph nodes. Occasionally, following
repeated exposures, fibrous tissue will form around
dead filariae that remain within lymph nodes. The fi-
brous tissue plugs up the lymphatic system, which re-
sults in swelling of the legs and genitals. The swollen

Figure 31-4
(edematous) areas become covered with thick, scaly
skin. This chronic disfiguring manifestation is called
elephantiasis because the extremities take on the ap-
pearance of elephant legs.
Diagnosis is made by the identification of microfilar-
iae in blood drawn at nighttime. (For poorly understood
reasons, very few organisms circulate during daylight
hours. This is called Nocturnal periodicity.) Diagno-
sis can also be made by identification of positive anti-
body titers via immunofluorescence.
Diethylcarbamazine is the agent used to treat ele-
phantiasis. Lymphatic damage may require surgical
correction.
Fig. 31-4. There are two ( Di) women named Ethyl in
this car: Di-ethyl-car. You will notice that there is an
elephant between Ethyl and Ethyl. You see, the drug
Diethylcarbamazine is used to treat the filarial infec-
tions caused by
Wuchereria bancrofti, Brugia malayi,
Loa boa,
and
Onchocerca volvulus.
These filariae chron-
ically infect the lymphatics, causing lymph obstruction,
giant swollen testicles, and elephant legs ("elephantia-
sis"). Thus the elephant between the Ethyls.
Dracunculus medinensis
(Guinea worm)
This very interesting tissue-invasive nematode lives as
a larval form inside intermediate hosts: African and
Asian freshwater copepods (tiny crustaceans). When a
person drinks water containing the microscopic crus-
taceans, the larvae penetrate the intestine and move
deep into subcutaneous tissue, where the adults develop
and then mate. The male is thought to die, but the female
grows over the course of a year to a size of 100 cm!!! She
then migrates to the skin and a loop of her body pokes out
and exposes her uterus. When the uterus comes into con-
tact with water, thousands of motile larvae are released.
Persons infected with
Dracunculus medinensis
will expe-
CHAPTER 31. HELMINTHS
256
rience allergic symptoms, including nausea, vomiting,
hives and breathlessness, during the larval release.
A common practice used to remove the worm involves
driving a small stick under the part of the worm's body
that is looped out of the skin. The stick is slowly twisted
each day to pull out the 100 cm
Dracunculus.
Cutaneous Larval Migrans
Also called creeping eruption, this intensely pru-
ritic, migratory skin infection commonly occurs in the
Southeastern U.S. The larvae of dog and cat hookworms
penetrate the skin and migrate beneath the epidermis.
As these larvae move (a few centimeters per day), an al-
lergic response is mounted, resulting in a raised, red,
itchy rash that moves with the advancing larvae.
The dog hookworm
Ancyclostoma braziliense
and
other species are responsible.
Human tissue-invasive nematodes such as the hook-
worm
(Necator americanus)
and
Strongyloides sterco-
ralis
can produce similar creeping eruptions.
Diagnosis is made by biopsy of the advancing edge of
the rash.
PLATYHELMINTHES
(Flatworms)
Flatworms do not have a digestive tract. There are
2 groups:
1) Trematodes, also known as flukes, include the
freshwater-dwelling schistosomes. Both male and fe-
male members exist and mate within humans (not in
the digestive tract, however). All flukes have a water
snail species as an intermediate host.
2) Cestodes, also known as tapeworms, live and
mate within the human digestive tract. Each tapeworm
has both male and female sex organs (hermaphrodites),
so individual tape worms can produce offspring by
themselves.
Trematodes
(Flukes)
Schistosoma
(Blood Flukes)
Schistosomal infections are extremely common
worldwide, second only to malaria as a cause of sickness
in the tropics. Schistosomes are found in freshwater.
They penetrate through exposed skin and invade the ve-
nous system, where they mate and lay eggs. Since the
eggs must reach freshwater to hatch, schistosomes can-
not multiply in humans.
Fig. 31-5. The 3 major
Schistosoma
species worldwide.

Figure 31-5
SCHISTOSOMES
The
Schistosoma
life cycle begins when their eggs
hatch in freshwater. Larvae emerge and swim until
they find a freshwater snail. After maturing within
these snails, the larvae are released and are now infec-
tious to humans. Mature schistosomal larvae (called
cercariae, which look like little tadpoles with oral
suckers on one end and a tail on the other), penetrate
through exposed human skin, and travel to the intra-
hepatic portion of the portal venous system. At this lo-
cation, the schistosomes rna`ture and mate. Depending
on the species, the pair of schistosomes will migrate to
the veins surrounding either the intestine or the blad-
der, where they lay their eggs. These eggs may enter the
lumen of the intestine or bladder, where they may be ex-
creted via feces or urine into a nearby stream or lake so
that they can continue their life cycle.
Interestingly, the adult worms in the venous system
are able to survive and release eggs for years, since they
are not killed by the immune system. It is thought that
schistosomes practice molecular mimicry (incorpora-
tion of host antigens onto their surface, which fools the
host's immune system into thinking that the schisto-
somes are NOT foreign).
However, cercariae (mature larvae) and eggs briskly
stimulate the immune system, and are responsible for
the systemic illness caused by this infection.
Clinical Manifestations
Schistosomiasis has 3 major disease syndromes that
occur sequentially: 1) Dermatitis as the cercariae pene-
trate a swimmer's skin, 2) Katayama fever as the
grown adults begin to lay eggs, and 3) Chronic fibrosis
of organs and blood vessels from chronic inflammation
around deposited eggs.
Upon penetration through the skin, patients tran-
siently develop intensely itchy skin (swimmer's itch)
and rash. Katayama fever follows 4-8 weeks later
with symptoms that can include fever, hives, headache,
weight loss, and cough. These symptoms may persist for
3 weeks. Lymph node, liver, and spleen enlargement
with eosinophilia are common.
When the schistosomes set up their home in the veins
surrounding the intestines or bladder, they begin re-
leasing eggs, many of which do not reach the feces or
CHAPTER 31. HELMINTHS
25 7
urine. Instead, these eggs are whisked off by the blood-
stream, where they are deposited in the liver, lung, or
brain. The immune system reacts against these eggs,
walling them off as granulomas. The deposition of eggs
in the venous walls of the liver leads to fibrosis, which
causes blockage of the portal venous system and a
backup of venous pressure into the spleen and mesen-
teric veins (portal hypertension). Any blood vessels or
organs that these eggs get stuck in can become in-
flamed, with formation of granulomas and ulcers. Pa-
tients may develop hematuria, chronic abdominal pain
and diarrhea, brain or spinal cord injury, or pulmonary
artery hypertension.
Diagnosis is based on the demonstration of eggs in
stool or urine samples and high eosinophil counts in the
blood. Control is directed at the proper disposal of hu-
man fecal waste and elimination of the snails that act
as the intermediate host.
Treatment
A group of quantum physicists got together to eradi-
cate this horrible disease. They wanted a drug that kills
all the flukes and tapeworms also. They succeeded so
Praise the quantum physicists! ( Note: This is a lie).
Praziquantel: This is truly a broad spectrum anti-
helminthic agent covering cestodes and trematodes
alike. When treating patients with praziquantel, don't
be surprised if there is an immediate exacerbation of
symptoms. The death of these schistosomes evokes a
vigorous immune response.
Cestodes
(Tapeworms)
Tapeworms are flatworms that live in the intestine of
their host. Since they lack a true digestive tract, they
suck up nutrients that have already been digested by
their host. Tapeworms are hermaphrodites (both male
and female organs in the same tapeworm), which en-
ables a single tapeworm to produce offspring. Humans
are usually the host of the adult tapeworm while other
animals may serve as intermediate hosts for the larval
stages.
Species
Geographic distribution
Resides in veins
surrounding
Deposits eggs
in:
Schistosoma japonicum
Eastern Asia
I ntestinal tract
Feces
Schistosoma mansoni
South America and Africa Intestinal tract
Feces
Schistosoma
haematobium
Africa
Bladder
Urine
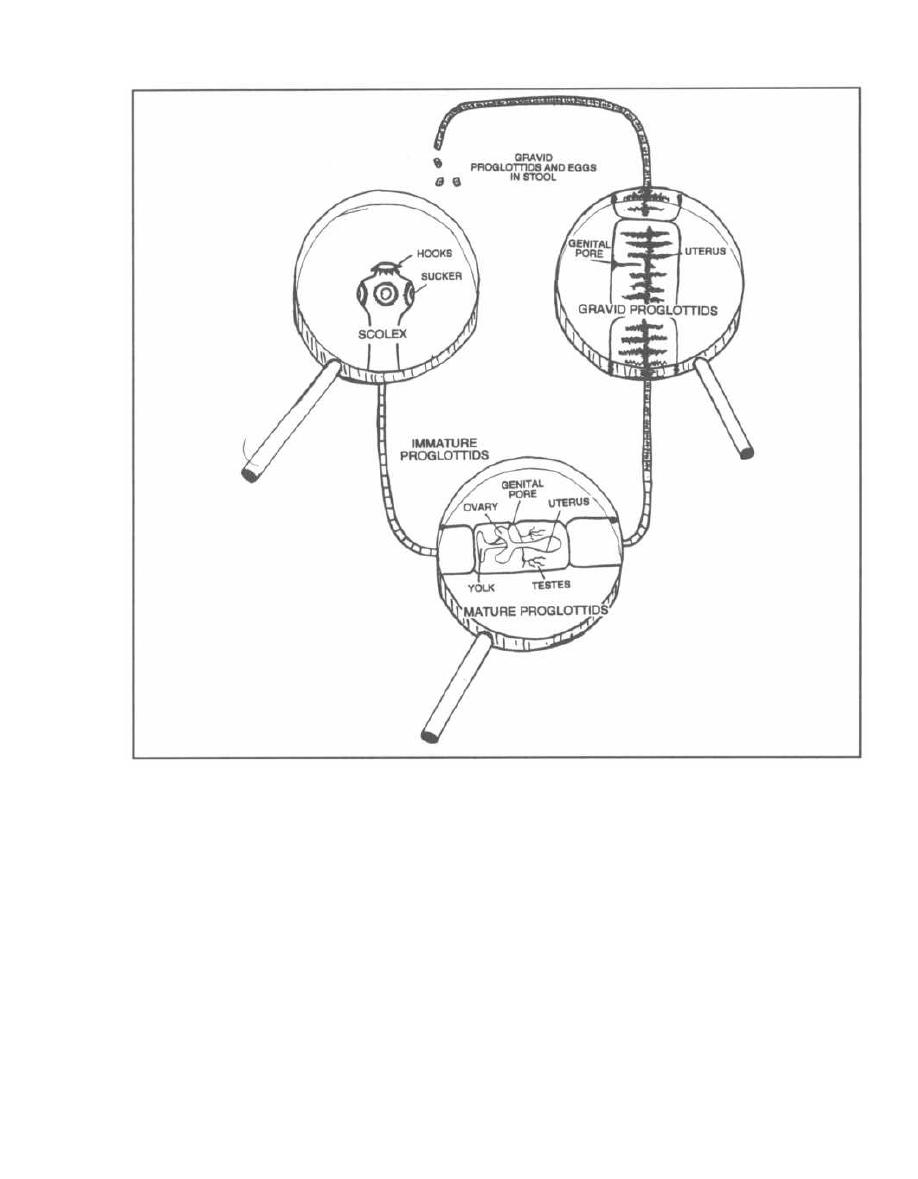
CHAPTER 31. HELMINTHS
Figure 31-6
Fig. 31-6. The tapeworm is long and flat (like a
typewriter ribbon) and consists of a chain of boxlike seg-
ments called proglottids. Let us examine the tape-
worm from its head down:
1) Scolex: The most anterior segment of the tape-
worm (the head), which has suckers and some-times
hooks.
2) Immature proglottids.
3) Mature proglottids have both male and female sex
organs.
4) Gravid proglottids contain fertilized eggs.
All of the tapeworm infections can be treated with
praziquantel or niclosamide.
258
Fig. 31-7. Niclosamide is an alternative agent to
praziquantel for treatment of tapeworm (cestode) infec-
tions. Picture a tapeworm wrapped around a nickel or
a nickel under tape.
Albendazole and praziquantel are used for the treat-
ment of
Taenia solium
larval cysticerci (see below).
Taenia solium
( Pork Tapeworm)
Humans acquire this infection by ingestion of under-
cooked pork infected with larvae. The pork tapeworm
attaches to the mucosa of the intestine via hooks on its
scolex and grows to a length of 2-8 meters. It releases
Figure 31-7
eggs in the human feces. When pigs graze on fields con-
taminated with the egg-infested human feces, they be-
come the intermediate host. The eggs develop into
larvae that disseminate through the intestine and into
muscle. In the animal's muscle tissue, the larvae de-
velop into another larval form, the cysticercus. The
cysticercus is a round, fluid-filled bladder with the lar-
val form within. When a human eats insufficiently
cooked pork muscle, the larval cysticercus converts to
the adult tapeworm in the intestine, and the cycle con-
tinues (see Fig. 31-8).
Infected individuals usually have minimal symp-
toms. Diagnosis is made by demonstration of proglottids
and/or eggs in a fecal sample.
Cysticercosis occurs when humans play the role of
the pig and ingest eggs rather than encysted lar-
vae. These eggs hatch within the small intestine, and
the larvae migrate throughout the body, where they
penetrate into tissue and encyst, forming cysticerci in
the human. Most commonly, the larvae encyst in the
brain and skeletal muscles (see Fig. 31-8).
CHAPTER 31. HELMINTHS
259
Cysticerci in the brain tend to cause more symptoms,
and this condition is called
Neurocysticercosis.
There are
usually 7-10 cysts in the brain, causing seizures, ob-
structive hydrocephalus, or focal neurologic deficits. The
cysts grow slowly and after 5-10 years begin to die and
leak their fluid contents. The antigenic contents cause lo-
cal inflammation and enhanced symptoms (more
seizures, meningitis, hydrocephalus, and focal deficits).
In endemic areas such as Mexico, Central and South
America, the Philippines, and SE Asia, Cysticercosis is
the most common cause of seizures.
The diagnosis of cysticercosis is made with the help of
a CAT scan or biopsy of infected tissue (brain or mus-
cle), both of which may reveal calcified cysticerci. Newer
serologic tests are also proving helpful in the diagnosis
of cysticercosis. Symptomatic disease, especially neuro-
cysticercosis, can be treated medically with dibendazole
or praziquantel.
Note that our immune system raises a red flag to
mark this invasion: the elevation of the eosinophil level
in the blood.
Taenia saginata
( Beef Tapeworm)
Taenia saginata
has the exact same life cycle as does
Taenia solium,
except that humans do not develop cys-
ticerci when they ingest eggs, as do the intermediate
hosts (cattle). For this reason, beef tapeworm infection
is relatively benign.
The beef tapeworm is acquired by the ingestion of lar-
val cysticerci in undercooked beef muscle. The adult
beef tapeworm develops and adheres (via suckers on its
scolex) to the intestinal mucosa, where it may reach a
length of over 10 meters and contain more than 2 thou-
sand proglottids.
Patients are usually asymptomatic, although they
may develop malnutrition and weight loss. Diagnosis
is made by identifying gravid proglottids and/or eggs
in feces.
Diphyllobothrium latum
(Fish Tapeworm)
The fish tapeworm can grow to 45 meters in length.
It is acquired by ingesting larvae in raw freshwater
fish. The life cycle involves the human and 2 fresh-
water intermediate hosts, a crustacean and a fish. The
adult tapeworms in the human intestine drop off their
gravid proglottids loaded with eggs. When the eggs
end up in water, they convert to a motile larval form,
which is then ingested by a crustacean, which is then
i ngested by a freshwater fish (trout, salmon, pike, etc.),
which is then ingested by a human-to end this long
sentence!
CYSTICERCI
LARVAE DEVELOP
INTO ADULT
TAPEWORMS IN
HUMAN INTESTINE
GRAVID
PROGLOTTIDS
AND EGGS
PASS IN FECES
HUMAN
HUMAN EATS
PORK WITH
u
CYSTICERCI
CYSTICERCOSIS:
HUMAN INGESTS EGGS.
HATCHED LARVAE
PENETRATE HUMAN MUSCLE,
BRAIN, LIVER, HEART, AND FORM
CYSTICERCI
PIGS INGEST
EGGS
PIG
CYSTICERCI
DEVELOP IN
PIG MUSCLE
1
LARVAE DEVELOP
AND MIGRATE INTO
PIG MUSCLE
Figure 31-8.
Pork tapeworm life cycle.
The large intestinal
Diphyllobothrium latum
tape-
worm provokes few abdominal symptoms, although it can
absorb vitamin
B12
to a significant degree. If vitamin
B,2
deficiency develops, anemia will occur. Diagnosis of in-
fection is made by identification of eggs in the feces.
Hymenolepis nana
(Dwarf Tapeworm)
This is the smallest tapeworm that infects humans
(only 15-50 mm in length), and it has the simplest life cy-
cle. There are NO intermediate hosts. Humans ingest
eggs that grow into adult tapeworms, and the adult tape-
worms pass more eggs that are again ingested by humans.
An infected patient will complain of abdominal
discomfort and occasionally nausea and vomiting.
Diagnosis is made by demonstration of eggs in a fecal
sample.
Echinococcus granulosus and
multilocularis
(Hydatid Disease, an Extra-intestinal Tapeworm
Infection)
Fig. 31-9. Dogs and sheep perpetuate the life cycle of
Echinococcusgranulosus
and the human is only a dead-
CHAPTER 31. HELMINTHS
260
end in the cycle.
Echinococcus
shares many similarities
with
Taenia solium,
with humans ingesting the eggs.
These eggs hatch in the intestine and develop into lar-
vae. After penetrating through the intestinal wall, the
larvae disseminate throughout the body. Most larvae
are concentrated in the liver, but larvae may also infect
the lungs, kidney and brain.
Each larva forms a single, round fluid-filled "hydatid"
cyst. These hydatid cysts can undergo asexual budding
to form daughter cysts and protoscolices inside the orig-
inal cyst. They can grow to 5-10 cm in size. Each cyst
may cause symptoms because it compresses the organ
around it (in the liver, lung, or brain). Humans are ex-
tremely allergic to the fluid within the cyst, and if the
cyst bursts, the allergic reaction may be fatal. While the
cysts of
Echinococcus granulosus
simply grow larger,
only to spread if they rupture, E.
multilocularis
cysts
undergo lateral budding and spread. These spreading
cysts can be misdiagnosed as a slowly growing tumor.
Diagnosis of the hydatid cyst employs similar tech-
niques as with
Taenia solium's
cysticerci cysts, using
CAT scanning and tissue biopsy. Only 10% of hydatid
cysts cause symptoms, and treatment of these is diffi-
cult. The best thing to do is to cut them out of the liver,
lung, or (yikes!) brain, but if the cyst fluid spills, out
will pour daughter cysts, protoscolices, and highly
EGGS IN DOG
'
SHEEP INGEST
STOOL
EGGS
LARVAE MIGRATE
INTO TISSUE
TAPEWORM
SHEEP
HYDATID CYSTS
FORM IN TISSUE
USUALLY
ONLY A SINGLE
HYDATID CYST
FORMS IN TISSUE
TISSUE WITH
CYSTS RUPTURE
CYST CAUSES
TAPEWORMS
INTO TISSUE
DAMAGE TO
INGESTED BY DOG
RELEASE TAPEWORMS
ORGANS
Figure 31-9.
Echinococcus life cycle.
allergenic fluid. Optional treatment usually starts with
treatment for months with albendazole to kill the cysts
(although this alone is rarely curative). The cyst is then
exposed surgically and some of the cyst fluid is carefully
aspirated. Saline, iodophors, or ethanol is next instilled
into the cyst to make sure the contents are dead. After
about 30 minutes the cyst is cut out.
When a hydatid cyst is inoperable due to a tricky lo-
cation or a poor surgical candidate, therapy with alber-
dazole is initiated and in some centers this is followed
by CAT scan guided fine needle injection of ethanol into
the cyst.
Since dogs and sheep perpetuate the cycle, efforts to-
ward control should target these animals. Dogs in graz-
ing countries should not be fed uncooked animal meat
and should be treated periodically with niclosamide.
Fig. 31-10.
Summary of the helminths.
Fig. 31-11.
Summary of the anti-helminths drugs.
26 1
References
Grove DI. Tissue Nematodes (Trichinosis, Dracunculiasis, Fi-
lariasis. In: Mandell GL, Bennett JE, Dolin R. editors. Prin-
ciples and Practice of Infectious Diseases. 4th edition. New
York: Churchill Livingstone 1995;2531-2537.
King HK. Cestodes (tapeworms). In: Mandell GL, Bennett JE,
Dolin R. editors. Principles and Practice of Infectious Dis-
eases. 4th edition. New York: Churchill Livingstone
1995;2544-2553.
Liu LX, Weller PF. Drug therapy: Antiparasitic drugs. N Engl
J Med 1996;334:1178-84.
Mahmoud AA. Trematodes (Schistosomiasis) and other
flukes. In: Mandell GL, Bennett JE, Dolin R. editors. Prin-
ciples and Practice of Infectious Diseases. 4th edition. New
York: Churchill Livingstone 1995;2538-2544.
Rosenblatt JE. Antiparasitic Agents. Symposium On Antimi-
crobial Agents-Part VII. Mayo Clin Proc 67:276-287, 1992.
Sanford JP, Gilbert DN, et al. Guide To Antimicrobial Therapy
1996. Antimicrobial Therapy, Inc, Dallas Texas; 1996.
CHAPTER 31. HELMINTHS
_
HUMANS INGEST
LARVAE MIGRATE
EGGS
INTO TISSUE
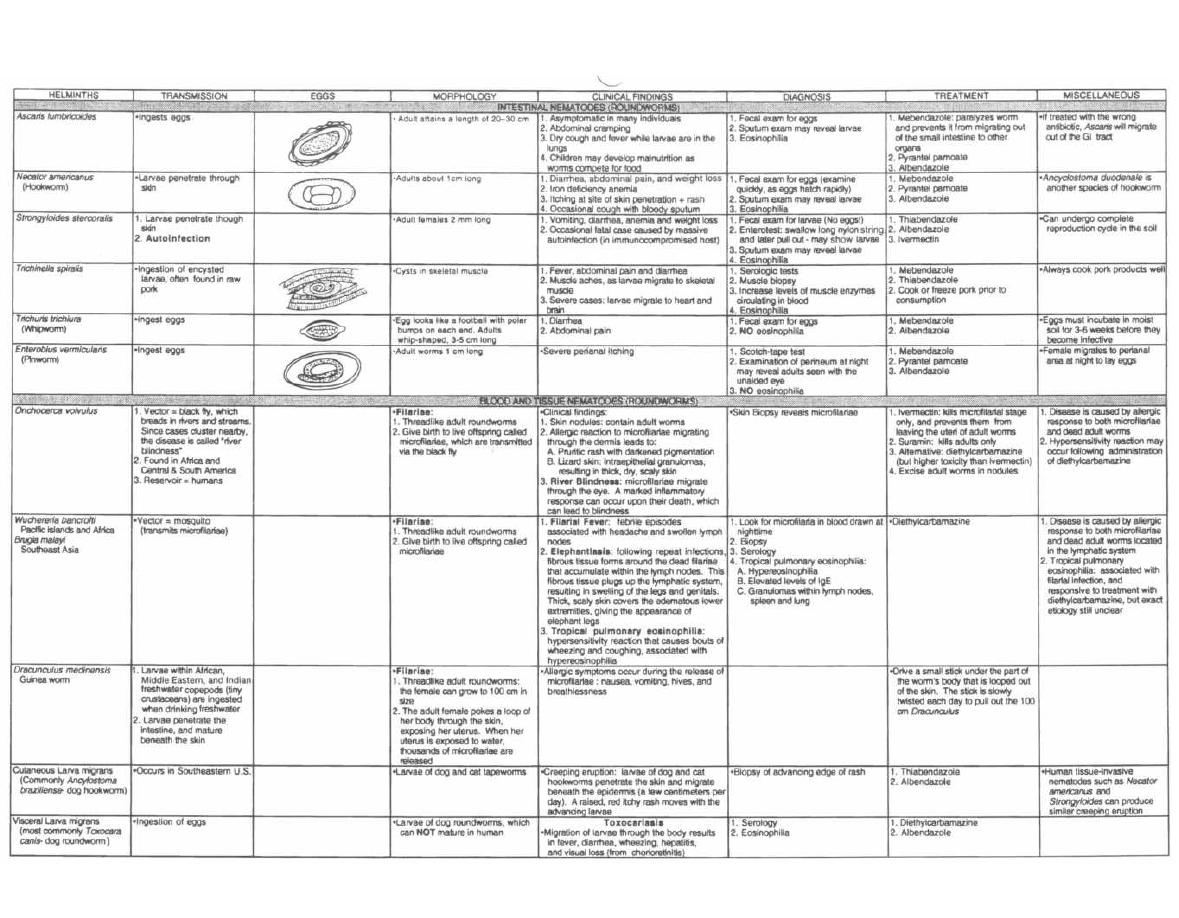
Figure 31-10 HELMINTHS
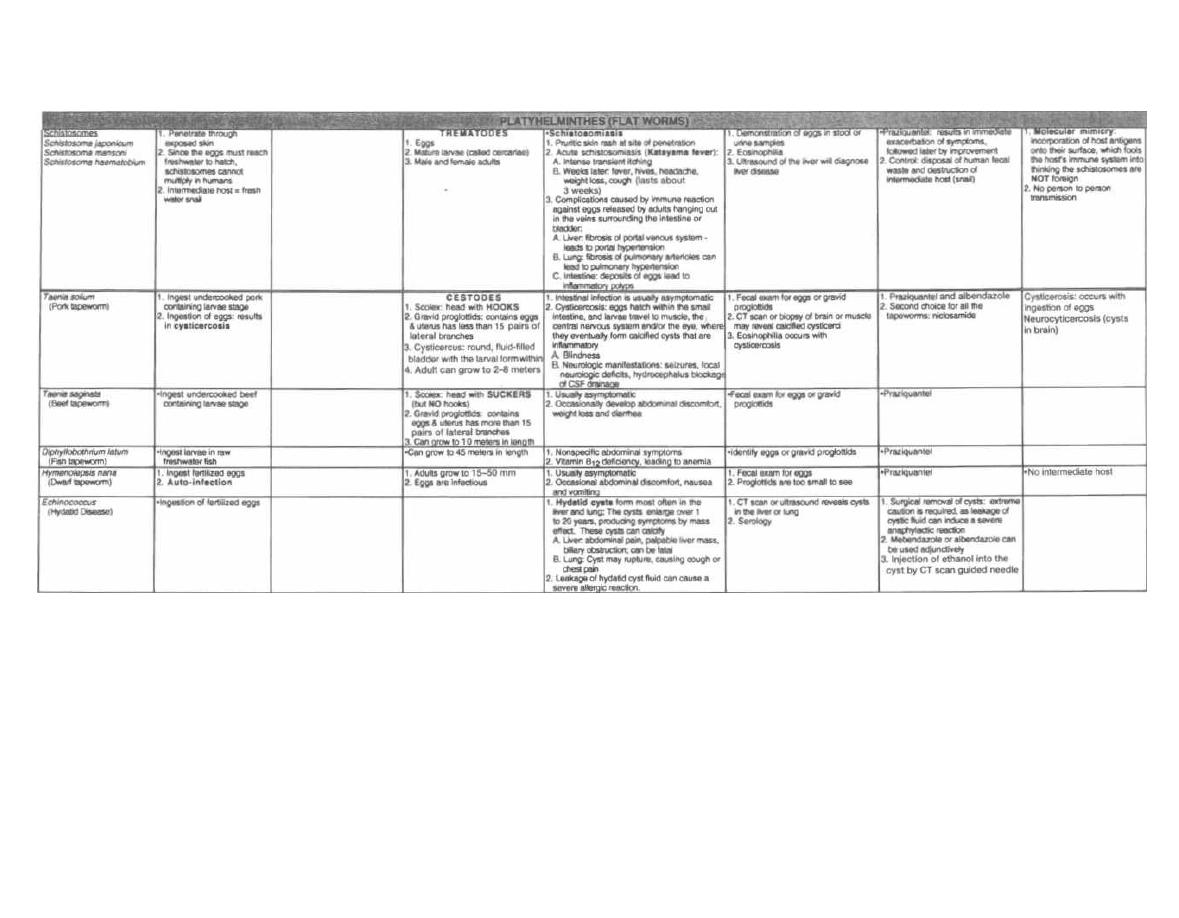
Figure 31-10 (continued)
M. Gladwin and B. Trattler,
Clinical Microbiology Made Ridiculously Simple
OMedMaster
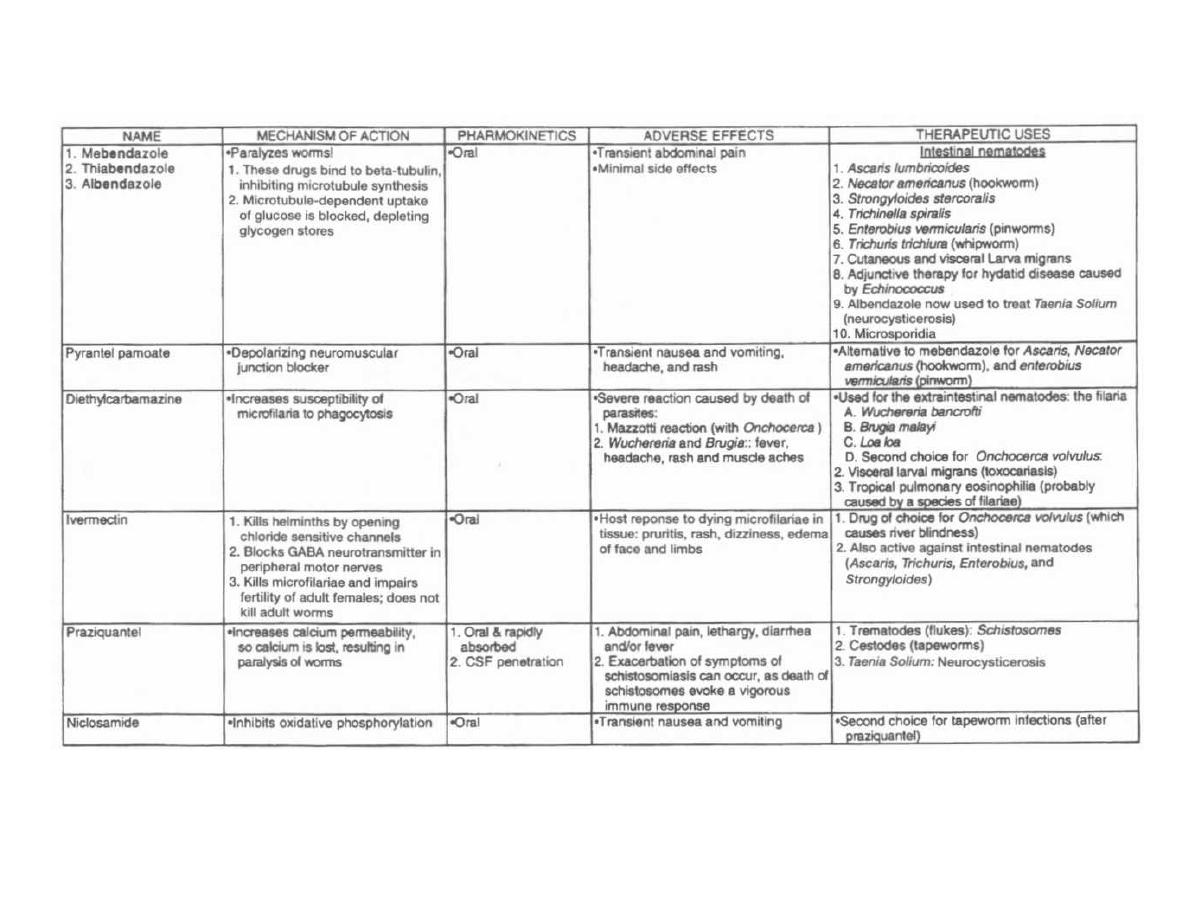
Figure 31-11
ANTI-HELMINTHS DRUGS
M. Gladwin and B. Trattler, Clinical
Microbiology Made Ridiculously Simple
©MedMaste

PART V. VERY STRANGE CRITTERS
CHAPTER 32. PRIONS
( Proteinaceous Infectious Particles):
Mad carnivorous cows, shaking cannibals, and
a few good reasons to be a vegetarian. .
.
Introduction:
The fancy name for Prion diseases is the transmissible
spongiform encephalopathies (TSE); so named because
these diseases are transmissible and create spongiform
pathological changes in the brain resulting in en-
cephalopathy (i.e. causing brain damage). These diseases
are fatal neurodegenerative disorders of humans and
other animals. The best known animal diseases are
scrapie in sheep and goats, and bovine spongiform en-
cephalopathy (BSE or mad cow disease) in cattle.
Four human prion diseases have been identified so
far, all of which are VERY rare: Creutzfeldt-Jakob dis-
ease (CJD), kuru ("shivering"), Gerstmann-Straussler-
Scheinker disease (GSS), and fatal familial insomnia (F F1).
Prion diseases share the following characteristics:
•
Long incubation time (months to years)
•
Gradual increase in severity leading to death
within months of onset
•
No host immune response
•
Non-inflammatory process in the brain
•
Neuro-pathological findings may include:
•
Macroscopical examination is often normal.
•
Microscopical spongiform changes (small, appar-
ently empty, microscopic vacuoles of varying sizes
within the neural tissue), neuronal loss, and
amyloid plaques (a pathological proteinaceous
substance deposited between cells) with accumu-
lation of the prion protein (PrP).
Prion diseases are unique in being both inherited and
infectious. There is also a sporadic manifestation with
no obvious inherited or infectious etiology. However,
neural tissue from individuals affected by either inher-
ited or sporadic (as well as infectious) form of prion dis-
eases is infectious!
The Infectious Agent and Etiology:
The nature of the infectious agent is still under investi-
gation and debate. Briefly, two main theories are debated:
The protein-only hypothesis and the viral hypothesis. Ad-
265
vocates for the protein-only theory find that the infectious
agent consists only of prion proteins with little, if any, nu-
cleic acid, whereas advocates for the viral theory argue
that it remains possible for prions to contain extremely
small amounts, or protected, nucleic acid.
Laboratory data indicate that the prion protein (PrP)
is the major (if not exclusive) component of prions. In
short, prions are resistant to agents that modify nucleic
acids but susceptible to agents that modify proteins.
The protein-only hypothesis is the most widely ac-
cepted theory today.
If prions do not contain nucleic acids, how can they be
infectious? How do they replicate? What about the ge-
netic code dogma? Isn't it heretical to suggest that an
agent without nucleic acids can replicate?
Well, it turns out that PrP is encoded by an endogenous
gene (for humans located on chromosome 20), but exists
in two conformational isoforms, that is PrP has two dif-
ferent (secondary and tertiary) protein structures:
•
The cellular isoform of PrP is constitutively ex-
pressed by normal animals, primarily in the brain
and is called PrPC for cellular. The function of PrPC
remains to be clarified.
•
The disease associated isoform of PrP is called PrP&
for scrapie.
What's the difference between the normal PrPC and
PrPSC? Again, the conformation (that is structure) of
PrPSC is very different from PrPC, and is thought to be re-
sponsible for the development of disease: Aberrant me-
tabolism of PrPSC results in accumulation of the protein
and brain injury.
The conformational change of PrPC is thought to be
post-translational, and can apparently be induced by
the presence of PrPSC, maybe with the interaction of
other proteins. Thereby a small amount of PrPSC will
initiate a chain-reaction of conformational change of
PrPC into PrPSC (see Fig. 32-1).
This is the clue to the above raised question about
replication of the prions if the infectious particles do not
contain nucleic acids. The prions do not need the genetic
software in the infectious particle-it's already present
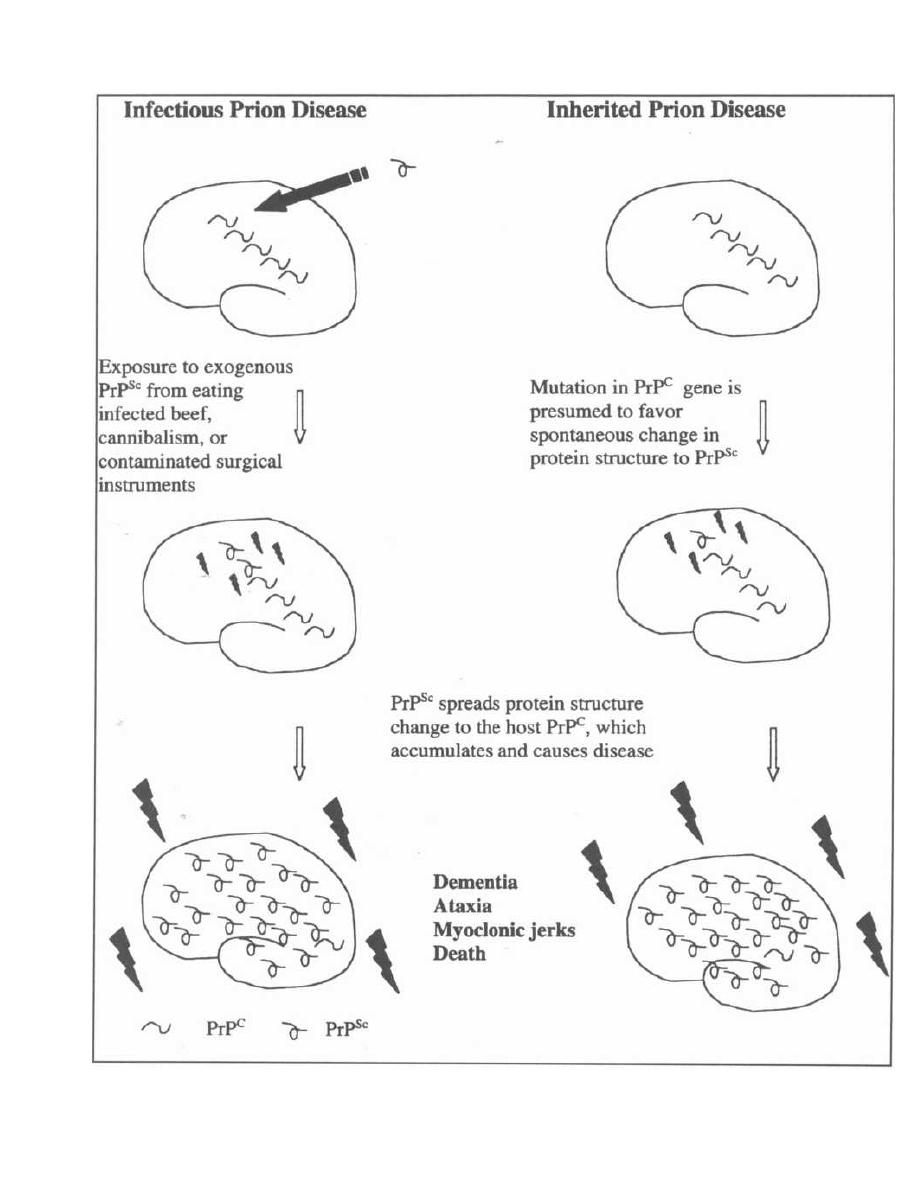
CHAPTER 32. PRIONS
Figure 32-1
266

in the host as part of the genome! The amino acid se-
quence of the "sick" isoform of PrP (PrPSC) accumulating
in a patient's brain is encoded by the PrP gene of that
particular individual!
Dr. Stanley B Prusiner proposed the name
prion
for
the agent causing transmissible spongiform en-
cephalopathy to emphasize the nature of these proteina-ceous infectious particles, and later concluded that the
infectious agent is PrP. He was awarded the Nobel
Prize in Physiology or Medicine in '97 for the discovery
of prions.
Basically three different etiologies are thought to be
involved relating to the nature of the disease:
•
Inherited: Mutations in PrP gene favoring sponta-
neous conformational change to PrPSC. The disease
is following an autosomal dominant pattern.
•
Infectious: Exogenous PrPSc inducing conforma-
tional change of host PrPC into PrPSC.
•
Sporadic: Unknown; probably spontaneous conver-
sion of wild-type PrP' or rare de novo mutations in
PrP gene leading to the conformational change
to PrPS~.
Transmission and Epidemiology:
Contaminated neural tissue has been shown to trans-
mit disease-even from species to other (i.e. crossing the
"species-barrier", e.g. sheep to cattle, cattle to human
etc). The route of transmission can be inoculation but
also
oral.
This has significant implications for the epi-
demiology of the disease.
Infectivity of other tissues and body fluids (e.g. blood)
are under investigation. The infectious particles are rela-
tively resistant to heat and many commonly used chem-
ical disinfectants as well as irradiation.
•
The bovine spongiform encephalopathy (BSE) epi-
demic among cattle is attributed to the practice of
feeding cattle (contaminated) sheep offal (the nice
wording is
meat and bone meal),
which basically is
the remainder of a butchered animal (bones etc), ac-
tually turning cattle into carnivores. This practice
has now been widely banned, although animal offal
are still fed to other livestock than cattle. Ulti-
mately, transmission of the disease from
mad cows
to humans causing a variant of Creutzfeldt-Jakob
disease (new variant Creutzfeldt-Jakob disease,
vCJD) has been established. Sale of meat on the
bone (e.g. T-bone steaks) has been banned to reduce
the contamination risk from neural tissue.
•
The kuru epidemic among the Fore population
of Papua New Guinea was probably transmitted
through ritualistic cannibalism (as a rite of mourn-
CHAPTER 32. PRIONS
26 7
ing the diseased). Kuru gradually disappeared after
the cessation of this practice. One can speculate
whether that epidemic originated with an index
case of sporadic Creutzfeldt-Jakob disease (sCJD).
•
Iatrogenic cases of Creutzfeldt-Jakob disease (CJD)
have been established through contaminated
(neuro) surgical instruments, dural and corneal
grafts and administration of cadaveric pituitary
hormones.
Clinical Presentation:
Until the
mad cow disease
epidemic in the UK in the
early 1990's causing human new variant Creutzfeldt-
Jakob disease (vCJD), prion diseases were widely un-
known to many physicians. The human prion diseases
are still VERY rare. Due to the large number of infected
cattle which have entered the human food-chain and the
possible long incubation time, it remains to be seen
whether the new variant Creutzfeldt-Jakob disease
(vCJD) will turn into an epidemic.
The prion diseases share many clinical features; all are
characterized by neurological signs and symptoms:
•
Rapidly progressive dementia
•
Psychiatric symptoms
•
Cerebellar symptoms (e.g. ataxia)
•
Involuntary movements (e.g. myoclonic jerks,
choreathetosis)
•
Ultimately fatal disease
Please refer to Fig. 32-2 for key features of individual
diseases.
Diagnosis:
•
The gold standard for diagnosis of prion diseases is
histopathologic examination and immunostaining
for PrPSC of brain tissue.
•
Cerebro-spinal fluid (CSF) is normal except that
mildly elevated protein levels may be seen. Eleva-
tion of certain proteins (14-3-3 and S100 proteins)
may occur in the cerebro-spinal fluid (CSF) as
well as in serum (S100 protein), but this is not spe-
cific for prion diseases and the utility remains to
be established.
•
Neuro-imaging tests (CT and MRI) may be normal,
and abnormal findings are generally not diagnostic.
•
Serial EEG can be very helpful in the diagnosis of
Creutzfieldt-Jakob disease. An abnormal pattern
(period sharp wave complexes) are ultimately seen
in more than 2/3 of Creutzfeldt-Jakob disease pa-
tients. However, this abnormality is generally NOT
seen in new variant Creutzfeldt-Jakob disease.
•
PrP is also present in lymphoreticular tissue, and
the utility of tonsil and spleen biopsies are under in-
vestigation as diagnostic tools.
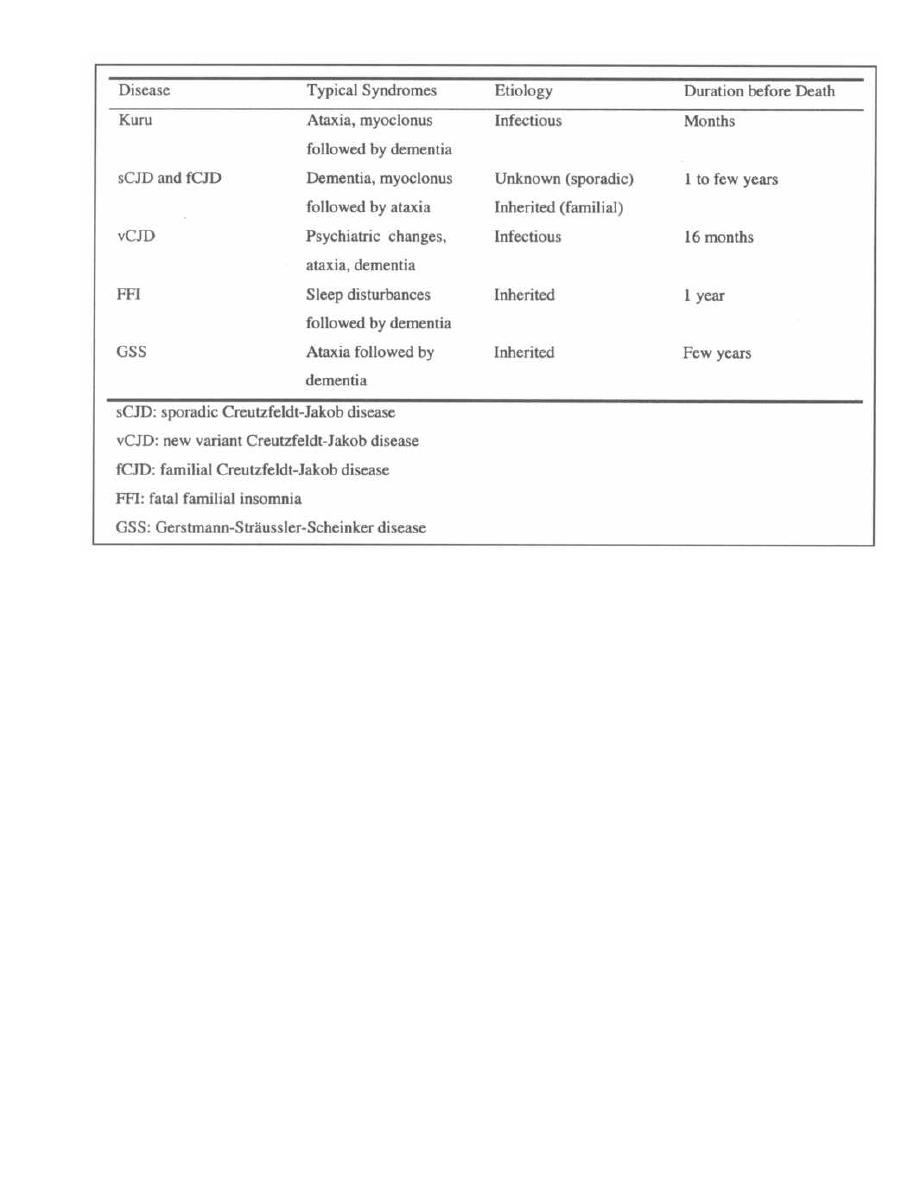
Figure 32-2
Treatment:
Unfortunately, this paragraph is going to be very short.
No curative treatment is currently available.
Recommended Reviews:
Belay ED. Transmissible Spongiform Encephalopathies in
Humans. Annu Rev Microbiol (1999) 53;283-314.
Johnson RT, Gibbs CJ. Creutzfeldt-Jakob Disease and Re-
lated Transmissible Spongiform Encephalopathies. NEJM
( 1998)339;1994-2004.
Prusiner SB. Prions. PNAS (1998) 95;13363-13383.
Tyler KL. Prions and Prion Diseases of the Central Nervous Sys-
tem (Transmissible Neurodegenerative Diseases). In Man-
dell et al. Principles and Practice of Infectious Diseases 5th
edition Churchill Livingstone (2000) pp. 1971-1980.
Bibliography:
Belay ED. Transmissible Spongiform Encephalopathies in
Humans. Annu Rev Microbiol 1999; 53:283-314.
Bruce ME et al. Transmissions to Mice indicate that °New
Variant" CJD is caused by the BSE agent. Nature 1997;
389:498-501.
CHAPTER 32. PRIONS
268
Chesebro B. Prion Protein and the Transmissible Spongiform
Encephalopathy Diseases. Neuron 1999; 24:503-506.
Gajdusek DC, Zigas V. Degenerative Disease of the Central
Nervous System in New Guinea. N Engl J Med 1957; 257:
974-978.
Gajdusek DC. Unconventional Viruses and the Origin and
Disappearance of Kuru. Science 1977; 197:943-960.
Harris DA. Prion Diseases. Nutrition 2000; 16:554-556.
Johnson RT, Gibbs CJ. Creutzfeldt-Jakob Disease and Re-
lated Transmissible Spongiform Encephalopathies. N Engl
J Med 1998; 339:1994-2004.
Prusiner SB. Novel Proteinaceous Infectious Particles cause
Scrapie. Science 1982; 216:136-144.
Prusiner SB. Molecular Biology and Genetics of Neurodegen-
erative Diseases caused by prions. Adv Virus Res 1992;
41:241-280.
Prusiner SB. Prions. PNAS 1998; 95:13363-13383.
Scott MR et al. Compelling Transgenic Evidence for Trans-
mission of Bovine Spongiform Encephalopathy Prions to
Humans. PNAS 1999; 96:15137-15142.
Tyler KL. Prions and Prion Diseases of the Central Nervous
System (Transmissible Neurodegenerative Diseases). In
Mandell et al. Principles and Practice of Infectious Diseases
5th edition Churchill Livingstone 2000; pp. 1971-1980.

DEVELOPMENT AND SPREAD OF
ANTIMICROBIAL RESISTANCE
With the current widespread overuse of antibi-
otics, we are forcing bacteria to genetically change to
survive. This antibiotic Darwinism has led to numer-
ous highly resistant strains of bacteria that now threaten
to create a Post-Antibiotic Era. Several factors are as-
sociated with emergence of resistance among organisms.
These
factors include:
1) Widespread, inappropriate use of broad-spectrum
antibiotics, especially in daycare centers and ICUs.
(e.g. treatment of viral illnesses with antibiotics).
2) Use of antibiotics in animal husbandry and fisheries
to prevent infection and increase animal growth.
3) Excessive use of antimicrobial preparations in
soaps and cleaning solutions in non-healthcare
facilities.
4) Increased numbers of immunocompromised pa-
tients requiring prolonged courses of antibiotics.
5) Prolonged survival of debilitated patients.
6) International travel promoting the movement of re-
sistant bacteria (e.g.
Mycobacterium tuberculosis).
7) Poverty leading to inadequate antibiotic usage be-
cause of the increasing expense of adequate an-
timicrobial therapy.
Fig. 33-1. describes setting-specific contributing factors
and resistant strains produced in more detail.
From examining antimicrobial resistance trends and
outbreaks, there are certain principles that continue
to hold true. First, given sufficient time and drug use,
antimicrobial resistance will emerge. There are no anti-
microbials to which resistance has not eventually ap-
peared. Second, the development of resistance is
progressive, evolving from low levels through interme-
diate to high levels. The exception to the rule is the di-
rect transfer of genetic information by plasmid or
transposon, which can result in immediate high level re-
sistance. Third, organisms that are resistant to one
drug are likely to become resistant to others. Cross-
resistance among a class of drugs or resistance to mul-
tiple classes of antibiotics are both possible (e.g.
Streptococcus pneumoniae). Fourth, once resistance ap-
pears, it is likely to decline slowly, if at all. Fifth, the use
of antimicrobials by any one person affects others in the
extended as well as the immediate environment.
Chapter 33. ONE STEP TOWARDS THE POST-ANTIBIOTIC ERA?
269
MECHANISM OF BACTERIAL
GENETIC VARIABILITY
Genetic variability is essential in order for microbial
evolution to occur. There are 3 basic mechanisms of
genetic variability leading to resistance among bacteria.
I1 Point mutations may occur in a nucleotide base
pair, which is referred to as microevolutionary
change. These mutations may alter the target site of
an antimicrobial agent, interfering with its activity.
2) Macroevolutionary change results in re-
arrangements of large segments of DNA as a
single event. These rearrangements may include
inversions, duplications, insertions, deletions or
transposition of large sequences of DNA from one
location of a bacterial chromosome to another.
3) Acquisition of foreign DNA carried by plasmids,
bacteriophages or transposable genetic elements (see
Chapter 3). These foreign elements give the organ-
ism the ability to adapt to antimicrobial activity.
MECHANISMS OF ANTIMICROBIAL
RESISTANCE
This genetic variability can be further separated into
more specific resistance mechanisms. These mecha-
nisms of resistance are as follows:
1) Enzymatic inhibition of antibiotics leading to an-
tibiotic inactivity.
2) Alterations of bacterial membranes to pre-
vent entry of antibiotics into bacteria.
3) Promotion of antibiotic efflux which actively
pumps the antibiotics out of the bacteria.
4) Alterations of bacterial protein targets which
make these targets unrecognizable to antibiotics.
Specific examples include:
•
Alterations of ribosomal target sites
•
Alterations of cell wall precursor targets
•
Alterations
of
critical enzymes
5) Bypass of antibiotic inhibition allowing bacte-
ria to find alternate pathways to survive when one
pathway is blocked by an antibiotic.
Enzymatic Inhibition
Enzymatic inhibition is one of the most common
modes of antimicrobial resistance. For example,
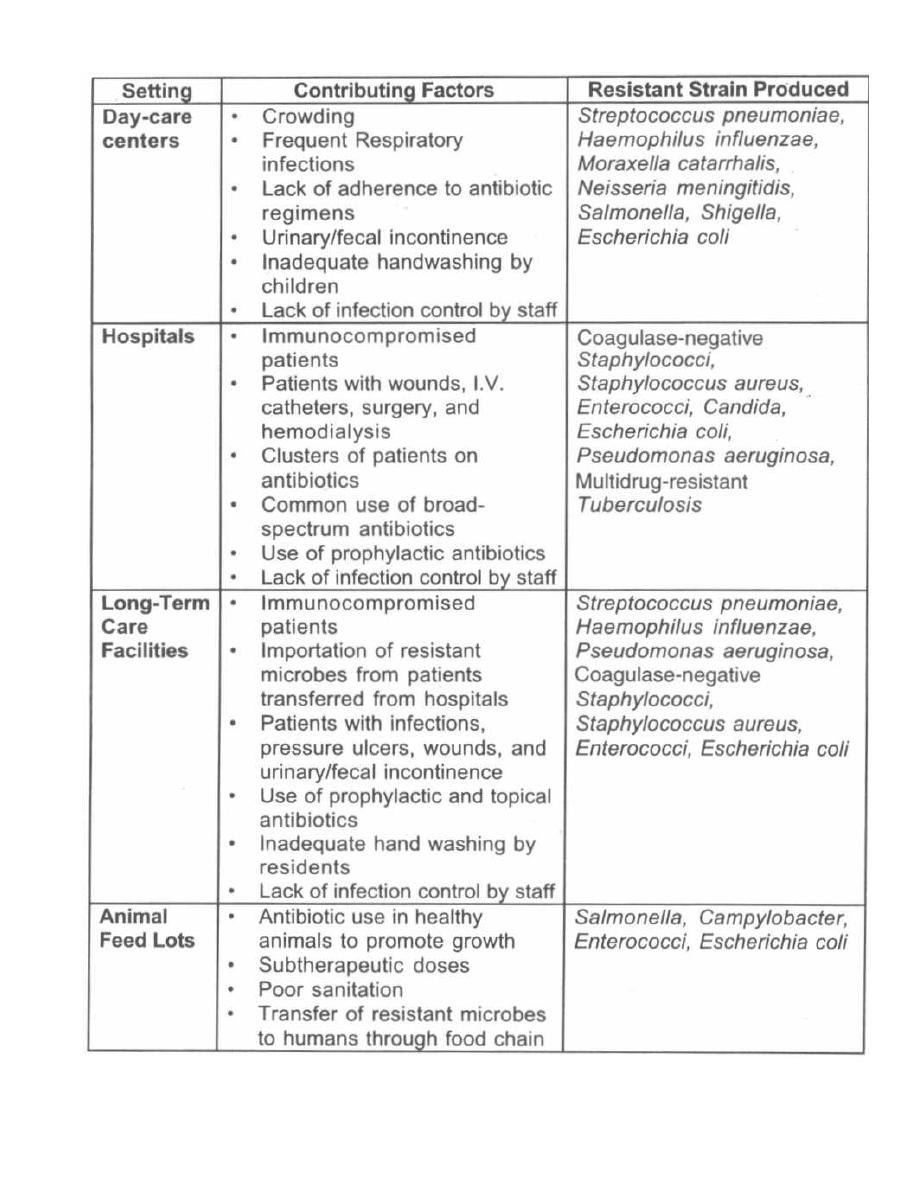
CHAPTER 33. ONE STEP TOWARDS THE POSTANTIBIOTIC ERA?
Figure 33-1

CHAPTER 33. ONE STEP TOWARDS THE POST-ANTIBIOTIC ERA?
Staphylococcus aureus' resistance to beta-lactam an-
tibiotics (e.g. penicillin) is due mainly to the production
of beta-lactamases, enzymes that inactivate these an-
tibiotics by splitting the beta-lactam ring. Enterococci
and gram-negative bacilli resistance to aminoglycosides
is also commonly due to modifying enzymes that are
coded by genes on plasmids or the chromosome.
Alterations of Bacterial Membranes
Outer Membrane Permeability: Many penicillins have
activity against gram-positive bacteria but not against
gram-negative bacteria because gram-negative bacteria
have a lipid bilayer that acts as a barrier to the penetra-
tion of antibiotics into the cell. Only when these penicillins
are able to get inside the cell are they able to work. Passage
of hydrophilic (water-soluble) antibiotics through this
outer membrane is facilitated by the presence of porins,
proteins that form water-filled diffusion channels through
which antibiotics can travel. Mutations resulting in the
loss of specific porins can occur and may lead to increased
resistance to penicillins.
Pseudomonas aeruginosa
resis-
tance to imipenem is a perfect example of this mechanism.
Inner Membrane Permeability: Aminoglycosides re-
quire active electron transport ("proton motive force")
which means that a positively charged aminoglycoside
molecule is "pulled" across cytoplasmic membranes of the
internal negatively charged cell. The energy generation or
the proton motive force that is required for substrate trans-
port into the cell may be altered in mutants resistant to
aminoglycosides.
Staphylococcus
resistant to aminoglyco-
sides is an example. These aminoglycoside-resistant or-
ganisms with altered proton motive force occur rarely, but
develop in the course of long-term aminoglycoside therapy.
Promotion of Antibiotic Efflux
The primary mechanism for decreased accumulation of
tetracycline is due mainly to active efflux of the antibiotic
across the cell membrane. Decreased uptake of tetracy-
cline from outside the cell also accounts for decreased
accumulation of tetracycline inside resistant cells. Tetra-
cycline resistance genes are generally inducible by
subtherapeutic concentrations of tetracycline which em-
phasizes the importance of adequate dosing.
Pseudomonas
aeruginosa
and
Staphylococcus aureus
are bugs that dis-
play this type of resistance to tetracycline. This system
may also represent a potential mechanism of resistance to
the newer quinolones, but has not been found to be com-
mon among quinolone-resistant clinical isolates.
Alterations of Bacterial Protein Targets
Alterations of Ribosomal Target Sites: Resistance
to a wide variety of antiribosomal agents, including
tetracyclines, macrolides, clindamycin, and the amino-
271
glycosides, may result from alteration of ribosomal
binding sites. Failure of the antibiotic to bind to its tar-
get sites on the ribosome disrupts its ability to inhibit
protein synthesis and cell growth. Ribosomal resistance
to streptomycin may be significant but is fairly uncom-
mon with gentamicin, tobramycin and amikacin. Ribo-
somal resistance can also be associated with decreased
intracellular accumulation of the drug. Examples in-
clude Staphylococcus
aureus
and Enterococci species re-
istance to macrolides.
Alterations of Cell Wall Precursor Targets: Resis-
tance of Enterococci to vancomycin has been classified
as A, B, or C based on levels of resistance. Class A re-
sistance is considered high level resistance and is asso-
ciated with the vanA gene. The vanA gene is carried on
a plasmid and encodes an inducible protein that is in-
volved in cell wall synthesis in E. Coli. These proteins
are responsible for synthesizing peptidoglycan precur-
sors that have a different amino acid sequence from the
normal cell wall peptidoglycan. This newly modified
peptidoglycan binds glycopeptide antibiotics with re-
duced affinity, thus leading to resistance to vancomycin
and teicoplanin. Classes B (vanB) and C (vanC) resis-
tance phenotypes are considered to have moderate
and low-level resistance respectively. The recent detec-
tion of decreased susceptibility to vancomycin among
Staph yloccus aureus is
also quite scary. Improvements
are being made in the ability of clinical laboratories to
characterize these resistant isolates.
Alterations of Critical Enzymes: Beta-lactam anti-
biotics inhibit bacteria by binding covalently to penicillin
binding proteins (PBPs) also called transpeptidases (see
Page 114) in the cytoplasmic membrane. These target pro-
teins are necessary for the synthesis of the peptidoglycan
that forms the cell wall of bacteria. Alterations of PBPs that
prevent successful binding can lead to beta-lactam resis-
tance. In gram-positive bacteria, resistance to beta-lactam
antibiotics may be associated either with a decrease in the
affinity of the PBP for the antibiotic or with a decrease in
the number of PBPs produced by the bacterium.
Bypass of Antibiotic Inhibition
Another mechanism for acquiring resistance to specific
antibiotics is by the development of auxotrophs, which
have growth factor requirements different from those
of the wild strain. These mutants require substrates
that normally are synthesized by the target enzymes,
and thus, if the enzyme is blocked and the substrates
are present in the environment, the organisms are
able to grow despite inhibition of the synthetic enzyme.
This is particularly concerning because the bacteria is
able to create additional pathways to meet growth
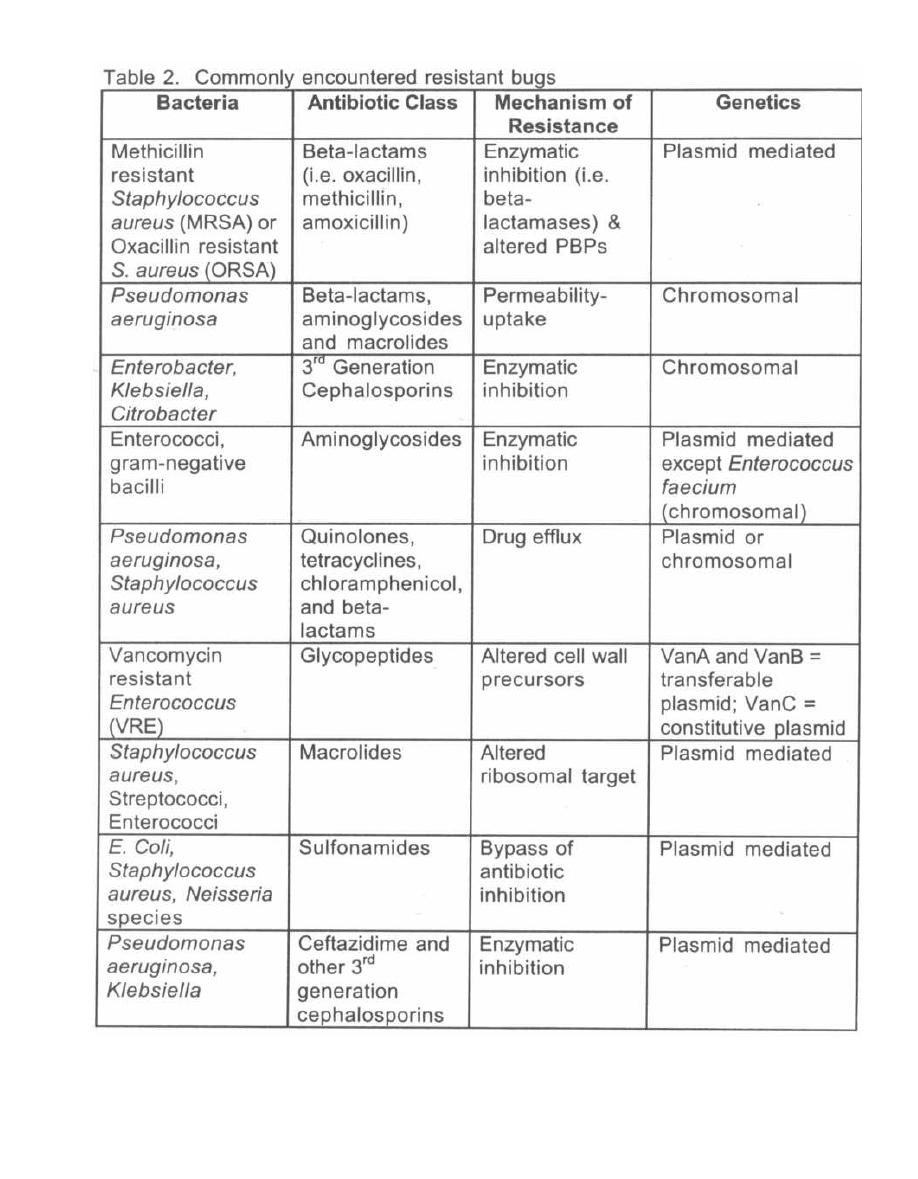
CHAPTER 33. ONE STEP TOWARDS THE POST-ANTIBIOTIC ERA?
272
Table 33-2

CHAPTER 33. ONE STEP TOWARDS THE POST-ANTIBIOTIC ERA?
requirements in response to a particular pathway being
blocked by the antibiotic. For example, "thymidine de-
pendent" bacteria like enterococci are able to utilize
exogenous supplies of thymidine for enzyme activity
and are thus highly resistant to trimethoprim which
blocks endogenous production of thymidine by bacterial
enzymes.
DECREASING ANTIMICROBIAL
RESISTANCE
In order to minimize antibiotic resistance in your pa-
tients you must employ these resistance management
approaches:
1) Withhold antibiotics i n situations where they
are not likely to benefit the patient for self-limited
viral infections such as "the common cold". Symp-
tomatic treatment and supportive measures are
the most appropriate care and antibacterial agents
are not indicated.
2) Use the narrowest spectrum antimicrobial
agent possible to treat an infection. For example,
a semisynthetic penicillin or even oral penicillin
would be a much better choice for treatment of a
staphylococcal infection than a broad spectrum
fluoroquinolone or cephalosporin. This works well
provided the organism is known or likely to be sus-
ceptible to the narrower spectrum antibiotic.
3) Base decisions about broadness of empiric
antibiotic coverage on the severity of illness.
For example, in the case of a patient who is clini-
cally stable and not at risk for significant morbidity
if a resistant pathogen is not treated immediately,
it may be appropriate to begin a narrow spectrum
agent while awaiting culture and susceptibility
data.
4) Emphasize prevention of infection through care-
ful hygiene, especially handwashing and other
measures to control the spread of pathogens. It
273
sounds really simple, but proper and adequate
handwashing by healthcare professionals can pre-
vent many cases of infection due to virulent and
antibiotic-resistant pathogens.
5) Utilize education to achieve therapeutic and pre-
ventative goals. Patients and families should be
counseled as to when antibiotics are needed, how
to take them correctly and for the proper dura-
tion. Education can also be used to foster earlier
detection of therapeutic failure, which may be
critical when treating patients who may be in-
fected with antibiotic-resistant pathogens. Our
communities must be cautioned against buying
cleaning products with antimicrobial properties as
well as using feed lot antibiotics.
References
Elliott TSJ, Lambert PA. Antibacterial Resistance in the In-
tensive Care Unit: Mechanisms and Management. British
Medical Bulletin
55:259-276, 1999.
Fridkin SK, Gaynes RP. Antimicrobial Resistance in Inten-
sive Care Units. Clinics in Chest Medicine
20:303-16, 1999.
Jones, RN. The Impact of Antimicrobial Resistance: Changing
Epidemiology of Community-acquired Respiratory-tract In-
fections. Am J Health-Syst Pharm
56:S4-S11, 1999.
Liu HH. Antibiotic Resistance in Bacteria: A Current and Fu-
ture Problem. RheumaDerm
1999; 59:387-396.
Mayer KH, Opal SM, Medeiros AA. Mechanisms of Antibiotic
Resistance. Basic Principles in the Diagnosis and Manage-
ment of Infectious Diseases
2000; 15:236-252.
Neely AN, Holder IA. Antimicrobial Resistance. Burns
25:17-24, 1999.
Reece SM. The Emerging Threat of Antimicrobial Resistance:
Strategies for Change. The Nurse Practitioner
24:70-86,
1999.
Virk A, Steckelberg JM. Clinical Aspects of Antimicrobial Re-
sistance. Symposium on antimicrobial agents. Mayo Clin
Proc
2000; 75:200-214.

3TC, 231
A subunit, 12
abacavir, 227, 232
ABCD, 155
ABLC, 155
abscess, 36
Acanthamoeba, 237
acid-fast stain, 106
acquired immunodeficiency syndrome, 192, 198
actinomycetes, 151
acute post-streptococcal glomerulonephritis, 25
acyclovir, 231
adenoviridae, 210
adhesins, 8
aerobes, 6
aerotolerant anaerobes, 7
aflatoxin, 151
African sleeping sickness, 246
AIDS related complex, 196
AIDS, 192, 198
albendazole, 235
alpha-hemolytic streptococci, 22
amantadine, 175, 229, 232
amastigotes, 245
Ambisome, 156
amikacin, 128
aminoglycosides, 128
aminopenicillins, 116
amoxicillin, 116
amphoteracin B, 144, 155
Amphotericin B colloidal dispersion, 155
Amphotericin B lipid complex, 155
amphotericin B deoxycholate, 156
ampicillin, 116
amprenavir, 228, 232
anaerobic gram-positive cocci, 65
Ancyclostoma braziliense,
252
anergy, 140
anthrax, 38
anti-C5a peptidase, 23
anti-viral medications, 224
antigenic drift, 173, 174
antimetabolites, 141
arboviruses, 214
ARC, 196
ArgyII-Robertson pupil, 93
artemether, 240, 250
artesunate, 250
Ascaris lumbricoides,
251
aspergilloma, 151
aspergillosis, 151
Aspergillus flavus, 151
athlete's foot, 145
INDEX
27,5
atovaquone, 250
atypical Mycobacteria, 106
Augmentin, 117
autotrophs, 7
azidodideoxythymidine (AZT), 225
azithromycin, 235
AZT, 202, 225
aztreonam, 122
B subunit, 12
B-cell lymphoma, 200
Babesiosis, 244
bacillary angiomatosis, 88
bacilli, 4
Bacillus, 38
Bacillus anthracis,
38
Bacillus cereus,
39
bacteremia, 12
Bacteroides fragilis, 64
Bacteroides melaninogenicus, 65
Balantidium coli,
248
Bartonella henselae,
88
Bartonella quintana,
88
basal body, 8
BCG vaccine, 104
beef tapeworm, 259
Bejel, 95
benznidazole treatment, 248
beta-hemolytic streptococci, 22
beta-lactam ring, 114
beta-lactamase inhibitors, 117
BK polyomavirus, 210
Blastomyces, 147
Bordet-Gengou medium, 70
Bordetella pertussis,
69
Borrelia burgdorferi,
96
Borrelia recurrentis,
98
botulism, 39
bovine spongiform encephalopathy, 265
Branhamella catarrhalis, 52
Brill-Zinsser disease, 87
Brucella, 75
Brugia malayi,
255
bubonic plague, 73
bunyaviridae, 216
Burkitt's lymphoma, 207
C carbohydrate, 22
CA proteins, 193
calcium alginate swab, 70
Caliciviridae, 219
California encephalitis, 216
Campylobacter jejuni,
62
Campylobacter pylori,
63
Candida albicans, 146, 150, 200

candidiasis, 150
capsid proteins, 193
capsid, 16, 161, 163
capsomer, 163
capsule b, 68
capsules, 9
carboxypenicillins, 117
carbuncle, 36
Carol Burnett, 89
caseous necrosis, 104
cat-scratch fever, 88
catalase, 6
catalase reaction, 22, 31
cefpodoxine, 120
ceftriaxone, 131
cell wall, 3
cellulitis, 36
cephalosporins, 117
cercariae, 257
cestodes, 256
Chagas' disease, 247
chagoma, 247
chemoheterotrophs, 7
chemotaxis, 8
chickenpox, 205
Chlamydia pneumoniae,
83
Chlamydia psittaci,
83
Chlamydia trachomatis,
79
Chlamydia, 78
chloramphenicol, 126
chloroquine, 243
cholera, 62
choleragen, 62
chromoblastomycosis, 146
chromotoid bodies, 234
ciprofloxacin (Cipro), 139
CKR5, 196
Cladosporium, 156
clarithromycin, 67, 127
clavulinic acid, 117
clindamycin, 127
clofazimine, 136
Clostridium, 38, 39
Clostridium botulinum
Clostridium difficile,
42
Clostridium perfringens,
42
Clostridium tetani,
40
cloxacillin, 116
clue cells, metronidazole, 69
CMV retinitis, 226
CMV, 200, 206
coagulase, 31, 32
cocci, 4
Coccidioides immitis,
200
cold agglutinins, 111
complement fixation test, 111
INDEX
276
condyloma latum, 91
cord factor, 102
core polysaccharide, 3
Coronaviridae, 219
Corynebacterium, 45
Corynebacterium diphtheriae,
45
Coxiella burnetti,
88
Coxsackie A and B, 218
creeping eruption, 256
Creutzfeldt-Jakob disease, 265
croup, 175
cryptococcosis, 149
Cryptococcus neoformans, 149, 200
Cryptosporidium, 235, 237
cutaneous larval migrans, 256
Cyclospora cayetanensis,
234, 249
cysticercus, 259
cytomegalovirus, 200, 206
d4T, 202, 227
dalfopristin, 27
Dane particle, 184
darkfield microscope, 5
ddC, 195, 202, 228, 231
ddl, 195, 202, 231
delaviridine, 228, 232
Dengue fever, 216
Dermacentor andersoni,
85
Dermacentor variabilis
,
85
dermatophytoses, 145
dicloxacillin, 116
didanosine, 195, 202, 225, 227, 231
dideoxycytidine, (ddC), 202, 228, 232
dideoxyinosine, (ddl), 202, 231
diethylcarbamazine treatment, 256
dimorphic fungi, 144
dimyristoylphosphatidylcholines, 155
dimyristoylphosphatidylglycerols, 155
Diphyllobothrium latum,
259
distearoylphosphatidylglycerol, 156
DNA viruses, 162, 166, 168
DNAases, 23
DPT, 41
Dracunculus medinensis,
256
Duffy antigens, 242
dwarf tapeworm, 256
Ebola virus, 221, 223
Echinococcus granulosus,
260
echoviruses, 218
edema factor, 39
efavirenz, 228, 232
Ehrlichia canis,
88
Ehrlichia chaffeensis,
88
Ehrlichiosis, 88
elementary body, 79
elephantiasis, 255
ELISA test, 201

EMB agar, 54
Embden-Meyerhof pathway, 7
endotoxins, 4, 12, 50
endospores, 10
Entamoeba histolytica,
234
enterics, 54
Enterobacteriaceae, 56
Enterobius vermicularis,
254
enterotest, 249
enterotoxins, 11, 32
Enterobacter, 57
enterovirus, 217
env, 194
envelope, viral, 164
epimastigotes, 245
Epstein-Barr virus, 200, 206, 207
ergosterol in fungi, 144
erythema chronicum migrans, 97
erythema infectiosum, 212
erythema marginatum, 24
erythema nodosum leprosum, 136
erythrocytic cycle, malaria, 242
erythrogenic toxin, 23
erythromycin, 127
Escherichia coli, 56
eucaryotes, 5
exfoliatin, 32
exo-erythrocytic cycle, malaria, 242
exosporium, 11
exotoxins, 11
extra cytoplasmic adenylate cyclase, 70
F protein, 175
F' plasmids, 20
F-plasmid, 19
F1 virulence factor, 73
facultative anaerobes, 6
facultative intracellular organisms, 11
famciclovir, 231
fatal familial insomnia, 265
fermentation, 7
Fifth disease, 212
filamentous hemagglutinin (FHA), 70
filariae, 254
fimbriae, 8
fimbrial antigens, 71
fish tapeworm, 259
Fitz-Hugh-Curtis syndrome, 83
flagella, 8
flatworms, 256
flaviviridae, 216
flesh-eating streptococcus, 24
flucloxacillin 116
fluconazole, 157
flukes, 256
fluoroquinolone antibiotics, 139
foscarnet, 231
INDEX
27 7
Francisella tularensis, 74
FTA-ABS test, 95
fungal antibiotics, 155
fungi, 144
furuncle, 36
fusin, 196
fusion protein, 175
Fusobacterium, 65
gag, 194
gametocytes, 242
gamma-hemolytic streptococci, 22
ganciclovir, 224, 225
Gardnerella vaginalis,
69
gas gangrene, 42
generalized transduction, 18
genciclovir, 231
Gerstmann-Straussler-Scheinker disease, 265
gentamicin, 128
Ghon complex, 105
Ghon focus, 105
Giardia lamblia,
234
glomerulonephritis, 25
gonorrhoeae, 51
gp120, 193
gp41, 193
gram stain, 1
gram-negative cell, 2, 5
gram-positive cell, 2, 4
griseofulvin, 155, 157
group antigen, 194
group B streptococci, 25
group D streptococci, 27
Guinea worm, 256
gummas, 92
H antigen, 55
H subunit, 12
Haemophilus ducreyi,
69
Haemophilus vaginalis,
69
Haemophilus, influenzae,
68
Hansen's disease, 107
hantavirus pulmonary syndrome, 216
heat-labile toxin, 39
heat-stable toxin, 39
helical symmetry capsids, 164
Helicobacter pylori,
63
helminths, 247
hemagglutinin, 172
hemolysins, 32
hemolytic uremic syndrome, 56
hemorrhagic fever, 216
hepatitis A virus, 180
hepatitis B virus, 182
hepatitis C virus, 188
hepatitis E virus, 188
hepatitis delta virus, 187
hepatitis, viral, 180

candidiasis, 150
capsid proteins, 193
capsid, 16, 161, 163
capsomer, 163
capsule b, 68
capsules, 9
carboxypenicillins, 117
carbuncle, 36
Carol Burnett, 89
caseous necrosis, 104
cat-scratch fever, 88
catalase, 6
catalase reaction, 22, 31
cefpodoxine, 120
ceftriaxone, 131
cell wall, 3
cellulitis, 36
cephalosporins, 117
cercariae, 257
cestodes, 256
Chagas' disease, 247
chagoma, 247
chemoheterotrophs, 7
chemotaxis, 8
chickenpox, 205
Chlamydia pneumoniae,
83
Chlamydia psittaci,
83
Chlamydia trachomatis,
79
Chlamydia, 78
chloramphenicol, 126
chloroquine, 243
cholera, 62
choleragen, 62
chromoblastomycosis, 146
chromotoid bodies, 234
ciprofloxacin (Cipro), 139
CKR5, 196
Cladosporium, 156
clarithromycin, 67, 127
clavulinic acid, 117
clindamycin, 127
clofazimine, 136
Clostridium, 38, 39
Clostridium botulinum
Clostridium difficile,
42
Clostridium perfringens,
42
Clostridium tetani,
40
cloxacillin, 116
clue cells, metronidazole, 69
CMV retinitis, 226
CMV, 200, 206
coagulase, 31, 32
cocci, 4
Coccidioides immitis,
200
cold agglutinins, 111
complement fixation test, 111
INDEX
276
condyloma latum, 91
cord factor, 102
core polysaccharide, 3
Coronaviridae, 219
Corynebacterium, 45
Corynebacterium diphtheriae,
45
Coxiella burnetti,
88
Coxsackie A and B, 218
creeping eruption, 256
Creutzfeldt-Jakob disease, 265
croup, 175
cryptococcosis, 149
Cryptococcus neoformans, 149, 200
Cryptosporidium, 235, 237
cutaneous larval migrans, 256
Cyclospora cayetanensis,
234, 249
cysticercus, 259
cytomegalovirus, 200, 206
d4T, 202, 227
dalfopristin, 27
Dane particle, 184
darkfield microscope, 5
ddC, 195, 202, 228, 231
ddl, 195, 202, 231
delaviridine, 228, 232
Dengue fever, 216
Dermacentor andersoni,
85
Dermacentor variabilis
,
85
dermatophytoses, 145
dicloxacillin, 116
didanosine, 195, 202, 225, 227, 231
dideoxycytidine, (ddC), 202, 228, 232
dideoxyinosine, (ddl), 202, 231
diethylcarbamazine treatment, 256
dimorphic fungi, 144
dimyristoylphosphatidylcholines, 155
dimyristoylphosphatidylglycerols, 155
Diphyllobothrium latum,
259
distearoylphosphatidylglycerol, 156
DNA viruses, 162, 166, 168
DNAases, 23
DPT, 41
Dracunculus medinensis,
256
Duffy antigens, 242
dwarf tapeworm, 256
Ebola virus, 221, 223
Echinococcus granulosus,
260
echoviruses, 218
edema factor, 39
efavirenz, 228, 232
Ehrlichia canis,
88
Ehrlichia chaffeensis,
88
Ehrlichiosis, 88
elementary body, 79
elephantiasis, 255
ELISA test, 201

EMB agar, 54
Embden-Meyerhof pathway, 7
endotoxins, 4, 12, 50
endospores, 10
Entamoeba histolytica,
234
enterics, 54
Enterobacteriaceae, 56
Enterobius vermicularis,
254
enterotest, 249
enterotoxins, 11, 32
Enterobacter, 57
enterovirus, 217
env, 194
envelope, viral, 164
epimastigotes, 245
Epstein-Barr virus, 200, 206, 207
ergosterol in fungi, 144
erythema chronicum migrans, 97
erythema infectiosum, 212
erythema marginatum, 24
erythema nodosum leprosum, 136
erythrocytic cycle, malaria, 242
erythrogenic toxin, 23
erythromycin, 127
Escherichia coli, 56
eucaryotes, 5
exfoliatin, 32
exo-erythrocytic cycle, malaria, 242
exosporium, 11
exotoxins, 11
extra cytoplasmic adenylate cyclase, 70
F protein, 175
F' plasmids, 20
F-plasmid, 19
F1 virulence factor, 73
facultative anaerobes, 6
facultative intracellular organisms, 11
famciclovir, 231
fatal familial insomnia, 265
fermentation, 7
Fifth disease, 212
filamentous hemagglutinin (FHA), 70
filariae, 254
fimbriae, 8
fimbrial antigens, 71
fish tapeworm, 259
Fitz-Hugh-Curtis syndrome, 83
flagella, 8
flatworms, 256
flaviviridae, 216
flesh-eating streptococcus, 24
flucloxacillin 116
fluconazole, 157
flukes, 256
fluoroquinolone antibiotics, 139
foscarnet, 231
INDEX
27 7
Francisella tularensis, 74
FTA-ABS test, 95
fungal antibiotics, 155
fungi, 144
furuncle, 36
fusin, 196
fusion protein, 175
Fusobacterium, 65
gag, 194
gametocytes, 242
gamma-hemolytic streptococci, 22
ganciclovir, 224, 225
Gardnerella vaginalis,
69
gas gangrene, 42
generalized transduction, 18
genciclovir, 231
Gerstmann-Straussler-Scheinker disease, 265
gentamicin, 128
Ghon complex, 105
Ghon focus, 105
Giardia lamblia,
234
glomerulonephritis, 25
gonorrhoeae, 51
gp120, 193
gp41, 193
gram stain, 1
gram-negative cell, 2, 5
gram-positive cell, 2, 4
griseofulvin, 155, 157
group antigen, 194
group B streptococci, 25
group D streptococci, 27
Guinea worm, 256
gummas, 92
H antigen, 55
H subunit, 12
Haemophilus ducreyi,
69
Haemophilus vaginalis,
69
Haemophilus, influenzae,
68
Hansen's disease, 107
hantavirus pulmonary syndrome, 216
heat-labile toxin, 39
heat-stable toxin, 39
helical symmetry capsids, 164
Helicobacter pylori,
63
helminths, 247
hemagglutinin, 172
hemolysins, 32
hemolytic uremic syndrome, 56
hemorrhagic fever, 216
hepatitis A virus, 180
hepatitis B virus, 182
hepatitis C virus, 188
hepatitis E virus, 188
hepatitis delta virus, 187
hepatitis, viral, 180

herpes simplex, 200, 204
herpes zoster, 200
heterophil antibody, 207
heterotrophs, 7
Hfr cell, 19
HHV-8, 200, 207, 208
Histoplasma capsulatum,
200
Histoplasma, 147
HIV, 192
HIV prophylaxis, 229
HLA-DR (1 + 4), 98
hookworm, 249
Hutchinson's teeth, 93
hyaluronidase, 23, 32
hydatid disease, 260
Hymenolipis nana, 260
hyphae, 144
hypnozoites, 242
icosahedral symmetry capsids, 163
IgA1 protease, 50
imidazoles, 157
imipenem, 120
impetigo, 36
inclusion conjunctivitis, 81
India ink stain, 10, 144
indinavir, 202, 228, 232
influenza, 173
initial body, 79
interferon, 230, 233
interferon alpha, 187
interleukin-1, 12
interleukin-2, 229
Intralipid, 156
isoniazid, 133
Isospora,
235, 237
Isospora, 233
itraconazole, 155, 157
ivermectin treatment, 255
Ixodes ticks, 96
Jarisch-Herheimer phenomenon, 95
JC polyomavirus, 210
jock itch, 145
K antigen, 55
kala-azar, 246
Kaposi's sarcoma, 200
Katayama fever, 257
ketoconazole, 144, 157
Klebsiella pneumoniae,
57
Koplik's spots, 176
L subunit, 12
lamivudine, 187, 195, 202, 227, 231
Legionella pneumophila,
72
Legionnaires' pneumonia, 71
Leishmania, 245
leishmaniasis, 240
leonine facies, 109
INDEX
278
lepromin skin test, 109
leprosy treatment, 135
leprosy, 107
Leptospira, 99
lethal factor, 39
leukocidins, 32
lipase, 32
lipid A, 4
Liposomal amphotericin B, 156
Listeria monocytogenes, 45
Listeria, 45
lockjaw, 41
Loeffler's medium, 45
long terminal repeat sequences, 193
LPS, 3, 50
LTRs, 193
Lyme disease, 96
Lymphogranuloma venereum, 83
lysogenic conversion, 19
lysogenic immunity, 17
M protein, 22, 172
MacConkey agar, 54
mad cow disease, 265
MAI, 106, 200
malaria, 240
Marburg virus, 221, 223
measles virus, 176
mebendazole treatment, 254
mefloquine, 243
melarsoprol, 250
meningitis, 50
meningococcal disease, 50
merozoites, 242
methicillin, 116
methicillin-resistant staphylococcus aureus, 36
metronidazole, 234, 236
microaerophilic bacteria, 7
microfilariae, 254
Microsporidia,
235
MIP1-Alpha, 196
MIP1-Beta, 196
MMR vaccine, 175
molds, 144
molecular mimicry, 257
molluscum contagiosum, 209
mononucleosis, 206, 207
Monospot test, 207
mumps virus 175
murein lipoprotein, 2
mycelia, 144
mycobacteria, 5
Mycobacterium avium-intracellulare,
106, 110, 200
Mycobacterium avium-complex,
106, 110, 200
Mycobacterium chelonei, 110
Mycobacterium fortuitum, 110
Mycobacterium kansasii, 110

Mycobacterium leprae,
107
Mycobacterium marinum, 110
Mycobacterium scrofulaceum, 110
Mycobacterium tuberculosis,
102, 200
Mycobacterium ulcerans, 110
mycolic acid, 102
mycoplasma, 5
Mycoplasma pneumoniae, 111
mycoside, 102
mycotoxins, 151
myonecrosis, 42
Naegleria fowleri,
237
nafcillin, 116
NC proteins, 193
Necator americanus,
251
necrotizing fasciitis, 23, 24
nef, 194
Negri bodies, 220
Neisseria catarrhalis,
52
Neisseria gonorrhoeae,
51
Neisseria meningitidis,
50
nelfinavir, 228, 232
nematodes, 251
neomycin, 128
neuraminidase, 172
neuroaminidase inhibitors, 229
neurotoxins, 11
nevirapine, 202, 228, 232
new enteroviruses, 218
niclosamide treatment, 258
nifurtimox treatment, 248, 250
NNRTIs, 227
nocardia, 151
nocturnal periodicity, 256
Norwalk virus, 219
nucleocapsid protein, 172, 193
nystatin, 144, 155, 157
O antigen, 3, 55
0-specific side chain, 3
obligate aerobes, 6
obligate anaerobes, 7
omeprazole, 67
Onchocerca volvulus, 255
oncogenes, vira1190
onychomycosis, 145
opportunistic infections, 200
optochin sensitivity, 27
oral hairy leukoplakia, 200
Orthomyxoviridae, 172
oseltamivir, 229, 232
oxacillin, 116
p24, 193
papilloma virus, 209
papovaviridae, 209
parainfluenza virus, 175
paramyxoviridae, 175
INDEX
279
parasites, 230
Parvoviridae, 166, 212
Pasteurella multocida, 76
PCP, 200, 235
pelvic inflammatory disease, 51
penicillin, 114
penicillin G, 115
penicillin V, 115
penicillinase, 32, 115
pentamidine, 250
peptidoglycan layer, 1, 3
peroxidase, 6
pertactin, 71
Pertussis toxin, 69, 70
Phialophora, 146
picornaviridae, 217
pili, 8
pinta, 96
pinworm, 254
piperacillin, 117
pityriasis versicolor, 144
plasmodia, 240
platyhelminthes, 256
pleomorphic bacteria, 4
Pneumocystis carinii,
238, 239
Pneumocystis carinii
pneumonia, 200
pol, 194
poliovirus, 217
polyomavirus, 210
Pontiac fever, 71
porin, 3
pork tapeworm, 258
potassium iodide, antifungal, 158
Pott's disease, 106
poxviridae, 166, 209
PPD skin test, 104
praziquantel, 257
primaquine, 243
prions, 265
pristinomycins, 27
procaryotes, 5
proglottids, 258
progressive multifocal leukoencephalopathy, 210
promastigotes, 245
prophage, 17
protease, 193, 194
protease inhibitor, 228, 232
protective antigen, 39
protein A, 32
Proteus and Rickettsia, 57
Proteus mirabilis, 57
protozoa, 234
pseudomembrane, 45
pseudomembranous colitis, 127
pseudomembranous enterocolitis, 42
Pseudomonas aeruginosa,
63

Pseudomonas cepacia,
64
psittacosis, 83
pyrantel pamoate treatment, 254
pyrazinamide, 134
pyrimethamine, 250
Q fever, 88
Quellung reaction, 10, 27
quinine, 239
quinolone, 131
quinupristin, 27
Ranke complex, 105
RANTES, 196
reduviid bug, 247
relapsing fever, 98
respiratory syncytial virus, 175
reticulate body, 79
retroviruses, 191
rev, 194
reverse transcriptase, 190
Reye's Syndrome, 174
rhabdoviridae, 220
rheumatic fever, 24
rhinovirus, 217, 219
ribavirin, 187, 216, 230, 233
rickettsia, 83
Rickettsia akari, 86
Rickettsia prowazekii, 86
Rickettsia rickettsia,
84
Rickettsia tsutsugamushi,
87
Rickettsia typhi,
87
rickettsialpox, 86
rifampin, 134
Rift Valley fever, 216
rimantadine, 229, 232
risus sardonicus, 41
ritonavir, 228, 232
river blindness, 255
RNA viruses, 161,166
RNA-dependent RNA polymerase, 162
Rochalimaea henselae, 88
Rochalimaea quintana, 88
Rocky Mountain spotted fever, 84
rotavirus, 219
roundworms, 251
RP59500, 27
RPR tst, 93
RSV, 175
rubivirus, 214
rule of sixes, 93
saber shins, 93
saddle nose, 93, 109
Salmonella, 58
saquinavir, 202, 228, 232
saprophytes, 144
scalded skin syndrome, 32, 34
schistosomes, 256
INDEX
28 0
schizont, 242
scolex, 258
scotch tape test, 254
scrapie, 265
scrofula, 106
scrub typhus, 87
self-transmissible plasmid, 19
sepsis, 12
septic shock, 12
Serratia, 58
sex pilus, 9, 19
Shiga toxin, 58
Shigella, 58
shingles, 205
smallpox, 209
specialized transduction, 18
spectinomycin, 130
spirochetes, 5, 91
spores, 144
Sporothrix schenckii, 146
sporozoites, 242
sprotrichosis, 146
staphylococci, 31
Staphylococcus aureus, 26
Staphylococcus aureus, 31, 32
Staphylococcus epidermidis, 36
Staphylococcus saprophyticus, 36
staphylokinase, 32
stavudine, 202, 227, 231
sterile pyuria, 106
stibogluconate treatment, 250
streptococcal pharyngitis, 23
streptococcal toxic shock syndrome, 23, 24
streptococci, 22
Streptococcus bovis,
27
Streptococcus equinus,
27
Streptococcus pneumoniae,
27
Streptococcus sanguis, 26
Streptococcus viridans,
26
Streptococcus mutans, 26
streptokinase, 23
streptolysin O, 23
streptolysin S, 23
streptomycin, 128, 135
Strongyloides stercoralis, 251
subacute sclerosing panencephalitis, 178
sulbactam, 117
sulfatides, 102
superoxide dismutase, 6
suramin treatment, 250
Synercid, 27
syphilis, 91
T-cell death, 198
T-strain Mycoplasma, 111
tabes dorsalis, 92
Taenia Saginata, 259

Taenia solium, 258
tapeworms, 256
tat, 194
tazobactam, 117
tellurite agar, 45
tetanospasmin, 41
tetany, 41
tetracycline, 128
thalidomide, 136
Thayer-Martin VCN media, 51
thiabendazole treatment, 253
thrush, 150
ticaricillin, 117
Timentin, 117
tinea capitis, 145
tinea corporis, 145
tinea cruris, 145
tinea nigra, 144
tinea pedis, 145
tinea unguium, 145
TNF, 12
tobramycin, 128
togaviridae, 214
TORCH viruses, 205, 216
toxic shock syndrome toxin, 33
toxins, 11
Toxoplasma gondii, 238
tracheal cytotoxin, 70
trachoma, 80
transduction, 16
transformation, 16
transpeptidase, 114
transposons, 21
trematodes, 256
trench fever, 88
Treponema pallidum,
91
Treponema subspecies, 95
triazoles, 157
Trichinella spiralis,
253
Trichomonas vaginalis,
236
Trichuris trichiura,
253
trifluridine, 233
trimethoprim and sulfa, 141, 235
trismus, 41
trophozoite, 242
tropical spastic paraparesis, 191
Trypanosoma, 245
trypanosomes, 241
trypomastigotes, 245
tsetse fly, 241
INDEX
28 1
Tsutsugamushi fever, 87
tuberculosis, 102
tuberculosis treatment, 133
tularemia, 74
tumor necrosis factor, 12
typanosoma cruzi, 242
typhoid fever, 59
typhus, 86
Unasyn, 117
Ureaplasma urealyticum, 111
ureidopenicillins, 117
V virulence factor, 73
V3
loop, 201, 202
valacyclovir, 231
vancomycin, 140
vancomycin resistant enterococci, 27
varicella-zoster virus, 205
VDRL, 93
Vi antigen, 58
Vibrio cholera,
62
Vibrio parahaemolyticus,
62
vidarabine, 233
viral load, 198
viral replication, 166
VRE, 27
W virulence factor, 73
walking pneumonia, 111
warts, 210
wax D, 103
Weil's disease, 100
Weil-Felix reaction, 84
West Nile virus, 216
western blot test, 201
whipworm, 253
whooping cough, 70
wood tick 85
Wood's light, 145
Wuchereria bancrofti,
255
xenodiagnosis, 248
yaws, 96
yeast, 144
yellow fever, 216
Yersinia enterocolitica,
61
Yersinia pestis,
73
zalcitabine (ddC), 195, 202, 227, 231
zanamavir, 229, 232
ZDV, 194, 202, 225, 228, 231
zidovudine (ZDV), 194, 202, 225, 228, 231
zoster, 205
Zosyn, 117