
1
Drugs Acting On the Cholinergic System and the Neuromuscular Blocking Drugs
Objectives and intended learning outcomes:
The student should be able to
List the locations and types of acetylcholine receptors in the major organ systems.
Describe the steps in the synthesis, storage, release and fate of acetylcholine.
Classify Cholinomimetic and describe their actions, uses and adverse Effects.
Classify different muscarinic antagonists and describe their actions, uses, adverse effects and
contraindications.
Describe the effects of nicotine and the ganglion-blocking drugs.
Classify neuromuscular blocking drugs, describe their mechanisms of action, report their
clinical applications, state their adverse effects and interpret their interactions.
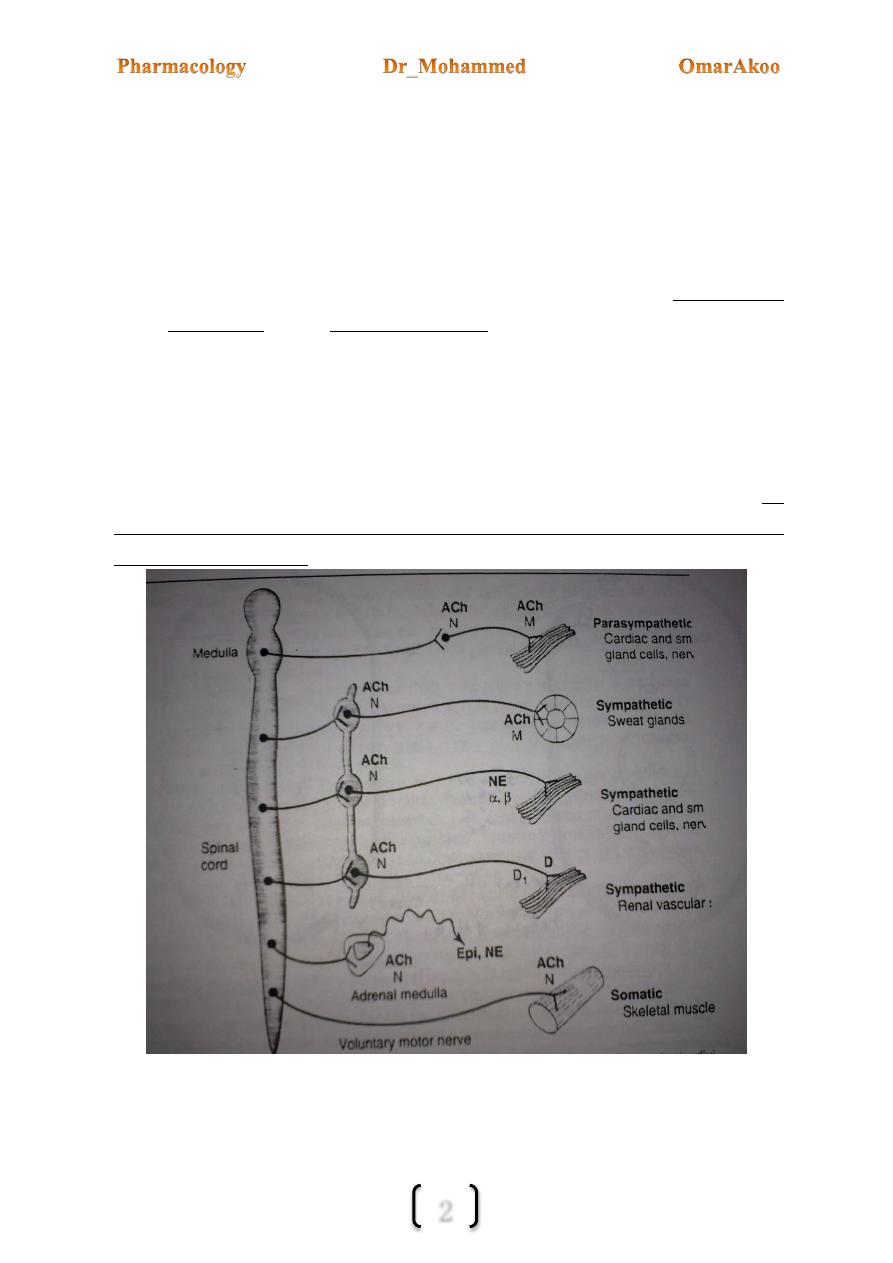
The autonomic nervous system (ANS) has two parts: the sympathetic and the
parasympathetic.
Now consider the following about the ANS:
1-Spinal roots of origin:
The parasympathetic fibers originate from the 3
rd,
7
th
, 9
th
& 10
th
cranial n. and from S2-S4
segments of the spinal cord, while the sympathetic preganglionic fibers originate in the
thoracic T1-T12 & Lumber L1-L5.
2-Location of the ganglia:
In the PANS (parasympathetic ANS) the ganglia are close to the innervated organ (i.e. the
preganglionic fiber is long and the postganglionic fiber is short) the opposite is true for
SANS (sympathetic ANS) because most of the sympathetic ganglia are located in the
paravertebral chains that lie along the spinal column.
2
3- Innervations of organs:
a) The motor efferent portion of the PANS is one of the motor pathways for
transmission of information from CNS to the effector tissues (smooth m., cardiac
m., exocrine glands).
b) Uninnervated receptors: some receptors that respond to autonomic transmitters and
drugs receive no innervation e.g. some muscarinic receptors on the endothelium of
blood vessels & some presynaptic receptors.
4- Neurotransmitters:
a. At the preganglionic synapse of the SANS & PANS the mediator is Ach.
b. At the postganglionic synapse the neurotransmitter in the PANS is Ach
while in the SANS it is norepinephrine (noradrenalin) with exception of the
thermoregulatory sweat gland (Acrine sweat gland) & some blood vessels to skeletal m.
where the mediator is Ach.
Diagram comparing some features of PANS & SANS with the somatic motor system
3
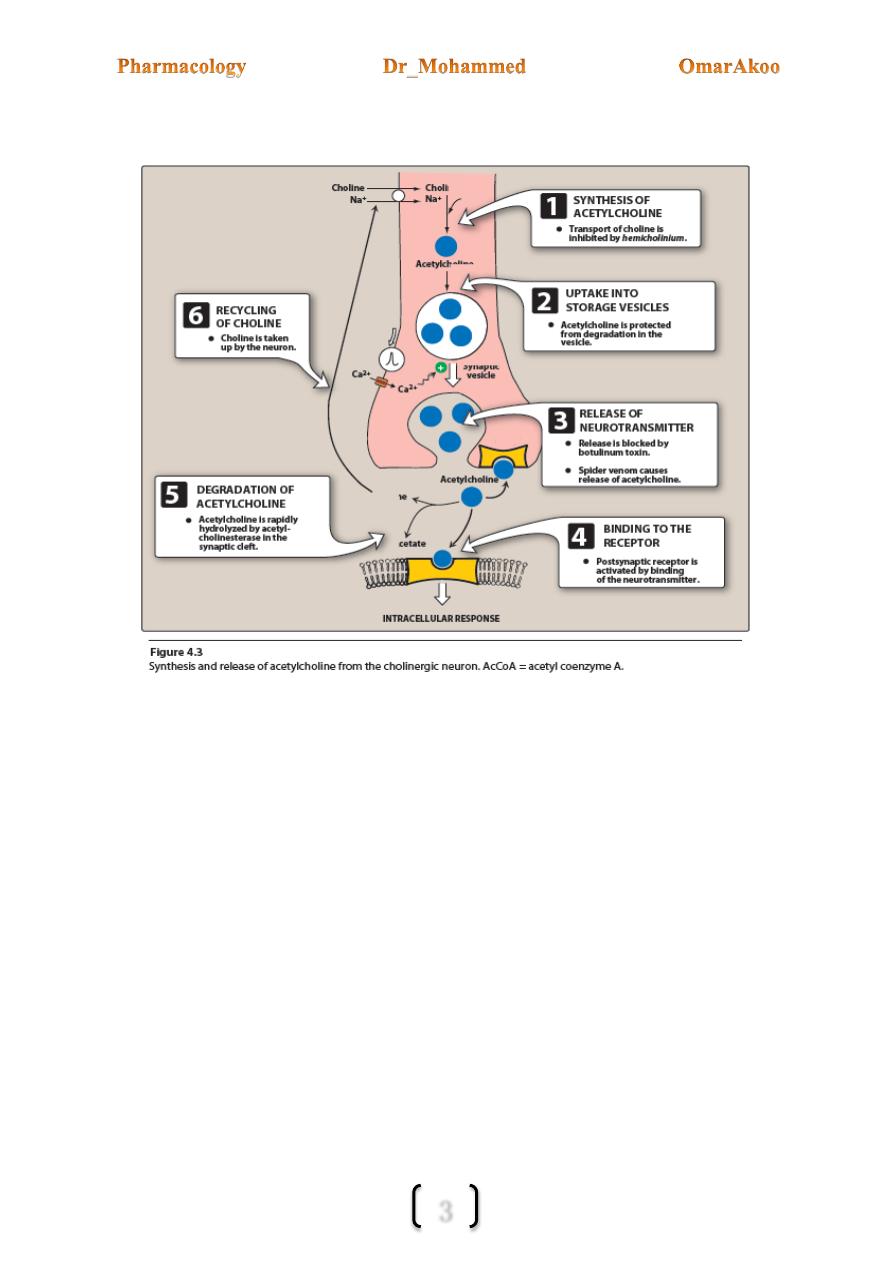
The synthesis, storage, release and termination of action of Ach:
Synthesis:
Ach is synthesized from acetyl co A and choline by the enzyme choline acetyl transferase.
The rate limiting step is probably the transport of choline into the nerve terminal. This
step is blocked by hemicholinium.
Storage:
Ach is actively transported into the vesicles for storage. This process is inhibited by
vesamicol

4
Release:
This occurs when an action potential propagated by the action of the voltage sensitive Na
channels arrives at the nerve ending.
The release of Ach requires entry of Ca++ ions (through Ca voltage gated channels which
become opened) and triggering interaction between several proteins associated with the
vesicle and nerve ending membrane (synaptobrevin, snap and others)
The interaction results in the fusion of the vesicular membrane and nerve ending
membrane and release of their contents into the synaptic space.
Botuloinum toxin blocks the release of Ach.
While (by contrast) black widow spider venom causes the release of all Ach stored in
the vesicles.
Binding to receptor:
Released Ach diffuses cross the synaptic space & bind either to postsynaptic receptor on the
target cells or to presynaptic receptor in the membrane of the neuron that released the Ach.
Termination of the action of Ach:
It is terminated in the synapse by metabolism of Ach to acetate & choline by the enzyme
acetylcholine esterase. Inhibition of those enzymes is important in the therapeutic effect of
many drugs.
Recycling of choline:
Choline is captured by Na coupled uptake system that transports the molecules back into the
neuron where it is acetylated and stored until released by a subsequent action potentiala (AP)
Cholinergic receptors (cholinoceptor):
These are subdivided into muscarinic & nicotinic receptors.

5
Muscarinic receptors (MRs):
These receptors are G coupled protein receptors.
Respond to muscarine as well as Ach.
The effects of activation of these receptors resemble those of postganglionic PANS
stimulation.
Muscarinic receptors are located primarily on autonomic effector cells including:
heart, vascular endothelium, smooth m., presynaptic nerve terminal and exocrine
glands.
Types: M1, M2, M3, M4, M5 but the first three types are the most important.
Nicotinic receptors (NRs):
These are ion cannel receptors.
Respond to nicotine (another Ach mimic) but not to muscarine.
There are two major subtypes of nicotinic receptors: (Nn) located in the ganglia and
(Nm) located at the neuromuscular end plate of skeletal m.
Nicotinic receptors are also found in the adrenal medulla and CNS
R
Location
Post R. Mechanism
M1
Nerve ending
Increase IP3, DAG cascade
M2
Heart , some nerve endings
Decrease CAMP, activate K channels
M3 Effector cells: smooth m., glands,
endoth.
Increase IP3,DAG cascade
Nn
ANS ganglia
Na/K depolarizing current (evokes AP)
Nm
N-M junction
Na/K depolarizing current

6
Mechanisms of Ach signal transduction (post receptor mechanisms)
Muscarinic mechanism:
Several mechanisms have been defined for muscarinic receptors:
1- The mechanism for M1 & M3 receptors:
When M1 & M3 are activated
(1) They undergo conformational change &
(2) Interact with a G protein which in turn
(3) Activates phospholipase C (a membrane bound enzyme) leading to
(4) Release of second messengers DAG (DiAcyl Glycerol) and IP3 (inositol
1,4,5,triphosphate).
DAG modulates the action of protein kinase C, an enzyme important in secretion while IP3
evokes the release of Ca from intracellular storage sites which results in contraction.
2- The mechanisms for M2 receptors:
a- Couples M2 to adenylyl cyclase through an inhibitory G protein which leads to
decrease cyclic AMP production.
b- Couples M2 receptor directly to K channel in the heart and elsewhere, muscarinic
agonists facilitate the opening of these channels.
Nicotinic mechanism:
The receptor is located on the channel protein that is selective to Na & K. When the receptor
is activated the channel opens and depolarization of the cell occurs (EPSP) as a direct result
of the influx of Na. Those receptors are present on the ganglionic cells (of both SANS &
PANS) & the neuromuscular junction.

7
Cholinergic drugs:
cholinergic drugs are of two types:
1-Direct acting agonists: acts directly on cholinoceptors.
2- indirect acting agonists (Anticholinesterase):acts by inhibiting the action of cholinesterase
accumulation of Ach in the synaptic space
Direct acting Cholinomimetic agonists:
These drugs mimic the action of Ach by binding directly to cholinoceptors.
Those agents are either:
1- Choline esters e.g. Ach, Methacoline, Carbacol and Bethanecol.
2- Naturally occurring alkaloids e.g. Muscarine, pilocarpine and Nicotine.
These drugs differ in their spectrum of action to muscarinic or nicotinic stimulation, e.g.
pilocarpine & Bethanecol preferentially bind to M.Rs, however direct acting drugs (as a
group) show little specify in their action.

8
Effects of direct acting cholinoceptors agonists
(Only the direct effects are indicated, haemostatic responses to these direct actions may be important)
Organ
Response
CNS
complex stimulatory effects e.g. nicotine (elevation of mood)
Eye
Sphincter m.
Ciliary's m.
miosis (constriction of the pupil)
contraction (accommodation) to near vision
Heart
SA node
AV node
Atria
Ventricles
heart rate (-ve chronotropy)
conduction velocity (-ve chronotropy) , R.P (refractory period)
contractile strength (-ve intropy) , R.P
small in contractile strength
Blood vessels
dilation via EDRF (endothelium derived relaxation factor) (NO)
Bronchi
bronchoconsriction
GIT
Motility
Sphincters
relaxation via ENS (enteric nervous system)
Urinary bladder
detrusor
trigon & sphincter
contraction
relaxation
Skeletal m,
(1) activation of N-M end plate
(2) contraction of m.
Glands
secretion of thermoregulatory sweat, lacrimal, bronchial, gastric and intestinal glands

9
NOTES:
Vasodilatation and decreased blood pressure is not evoked by PANS discharge.... why?
-Because it is mediated by the action of uninnervated muscarinic receptors found in blood
vessels which are stimulated by directly acting muscarinic (muscarinic agonists),normally
theses receptors have no function because Ach is never released into the blood in significant
quantities , stimulation of these receptors leads to the release of NO (EDRF) which causes
the vasodilatation.
Decreased blood pressure evokes a baroceptor reflex resulting in a strong sympathetic
discharge to the heart, thus the result may be tachycardia rather than bradycardia.
Another effect seen with directly acting drugs but not with PANS stimulation is
thermoregulatory sweating, this is a sympathetic-cholinergic effect.
Clinical uses:
We can predict the major clinical application of muscarinic from a consideration of organs
and diseases that benefit from an increase in cholinergic activity.
Ach:
Acetylcholine is quaternary Ammonium ester that is rapidly hydrolysed by acetylcholine
esterase & plasma cholinesterase (pseudo cholinesterase); Ach therapeutically is of no
importance. Why?
Because of: (1) its multiplicity of actions (leading to diffuse effects) and
(2) Its rapid inactivation by cholinesterase (5-30 sec.).
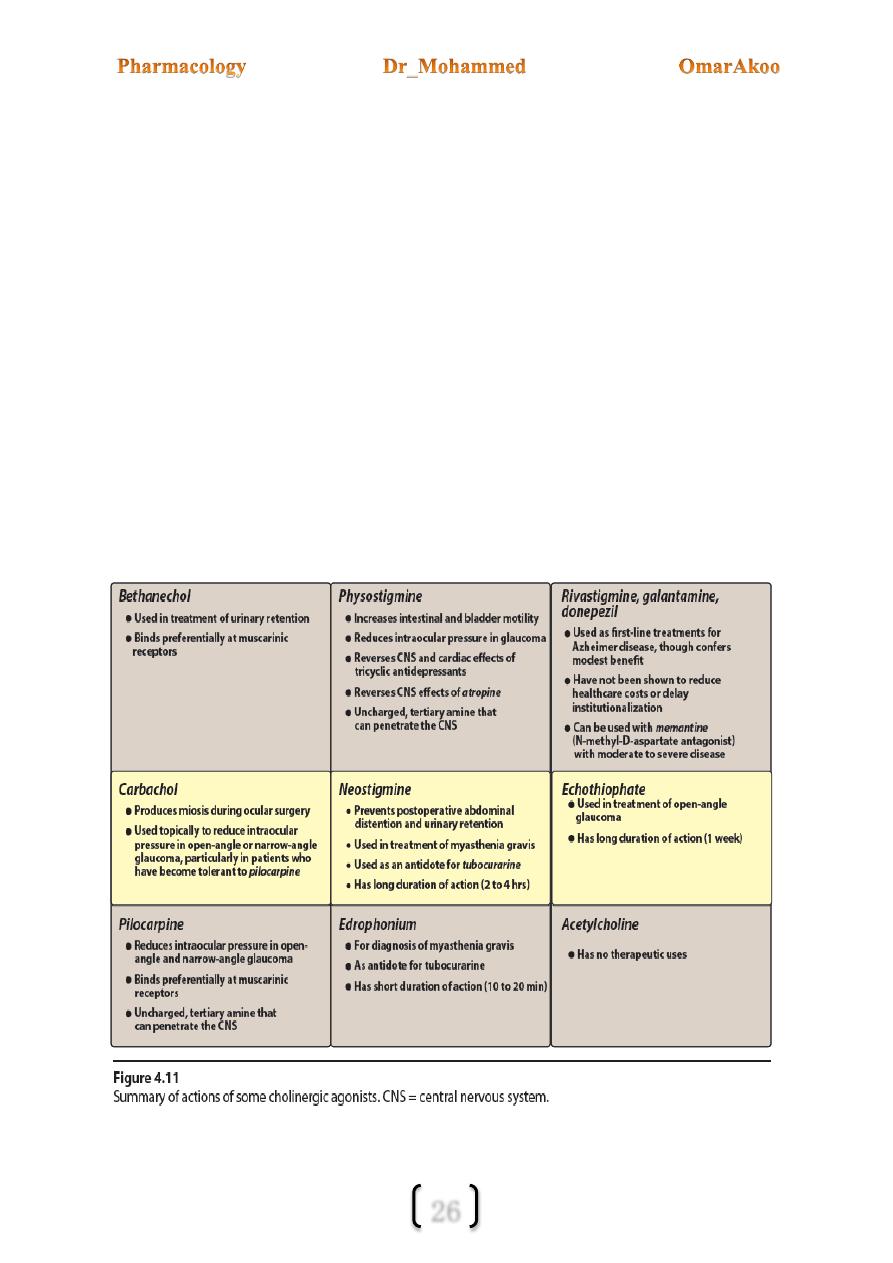
Bethanecol:
Structurally, it is related to Ach but it is resistant to hydrolysis by ChE (cholinesterase)
although it can be inactivated by other esterase.
It has strong muscarinic activity but no nicotinic action.
It is orally active,its duration of action is 30m – 2hrs.
Its action is mainly on the smooth m. of the GIT and bladder; therefore, it is used for post
operative & neurogenic ileus.

10
Carbachol:
Have both muscarinic & nicotinic actions
It is poor substance for ChE.
It causes the release of epinephrine from the adrenal medulla by its nicotinic action.
Duration of action is one hour.
It is used topically as a miotic agent & to decrease IOP (intraocular pressure).
Pilocarpine:
It is an alkaloid & is stable to hydrolysis by ChE.
It exhibits muscarinic activity.
Duration of action is 30m – 2 hrs.
It is primarily used in ophthalmology & it is the drug of choice in the emergency
lowering the IOP both in narrow angle and wide angle glaucoma.
Pilocarpine is extremely effective in the opening of trabecular meshwork around
schlemns canal causing an immediate drop of IOP as a result of increased drainage of
aqueous humor.
Pilocarpine can enter the brain and can cause CNS disturbances.
Toxicity:
The signs and symptoms of over dosage are readily predicted from the general
pharmacology of Ach
Toxicity includes CNS stimulation, miosis, spasm of accommodation,
bronchoconstriction, GIT and genitourinary smooth m. activity, secretion activity
of (sweat gland, airways and GIT) and vasodilatation.
Transient bradychardia followed by reflex tachycardia if the drug is administrated I.V.

11
Indirect acting agonists (Anticholinesterase):
Drugs that inhibit the activity of cholinesterase indirectly provide a cholinergic action by
prolonging the life time of Ach, produced endogenously at the cholinergic nerve terminal.
These drugs therefore lead to the accumulation of Ach in the synaptic space and can
produce a response at all cholinoceptor in the body, including both muscarinic and
nicotinic receptors of the ANS as well as the neuromuscular junction and the brain
Anticholinesterase can be subdivided into reversible AchE and irreversible AchE.
1-Reversible agents:
Are carbonic acid esters "carbamates"
Neostigmine is prototype.
2- Irreversible agents:
Are phosphoric acid esters "organophosphates"
Isoflurophate and echothiophate are prototypes.
A third class which has only one number:
Edrophonium: is an alcohol (not an ester) which is reversible with a very short duration of
action. Edrophonium binds reversibly to the active center of AChE, preventing hydrolysis
of ACh. It is rapidly absorbed and has a short duration of action of 10 to 20 minutes due to
rapid renal elimination.
It is used in the diagnosis of a myasthenia gravis, which is an autoimmune disease caused by
antibodies to the nicotinic receptor at NMJs. This causes their degradation, making fewer
receptors available for interaction with the neurotransmitter.

12
1- Reversible Anti ChE:
The carbamate residue is released by ChE over a period of 2-8 hours.
Physostigmine: is an alkaloid, it’s a tertiary amine (can enter and simulate the CNS)
Uses:
1- Because it increases intestinal and bladder motility thus it is used in case of atony of
bladder and paralytic ileus.
2- It is also used to treat over dose of drugs with anticholinergics action. Such as
Atropine, Phenothiazine, and TCA (tricyclic antidepressants)
3- Intraocular pressure (IOP) thus it is useful in glaucoma but pilocarpine is more
effective.
Adv. Effects:
CNS: may lead to convulsions. In over dose, bradychardia and neuromuscular (N-M)
paralysis may also occur.
Neostigmine: is a synthetic compound (doesn’t enter the CNS), its effect on the skeletal
muscles is more than Physostigmine because it also has direct action on skeletal m.
cholinergic receptors (nicotinic receptors of the N-M junction).
Uses
:
1- Post operative and neurogenic ileus (painful obstruction in the ileum and other part of
the intestine) and urinary retention.
2- Antidote for tubocurarine and other competitive N-M blockers.
3- Symptomatic treatment of myasthenia gravis (weakness in the muscles).

13
Pyridostigmine: it’s duration of action is (3-6 hours) which is longer than Neostigmine
(2-4 hours).
Uses:
in chronic management of myasthenia gravis.
Edrophonium: short duration of action (10-20 min).
Uses:
1- Used in diagnosis of myasthenia gravis and to differentiate myasthenia crisis from
cholinergic crisis.
2- Used as antidote to curare like drugs.
2- Irreversible Anti ChE:
A number of synthetic organophosphates have the capacity to bind covalently to ChE
forming an extremely stable phosphate complex with the enzyme.
The end result is increased Ach at all sites where it is released. Many of these drugs are
extremely toxic and were developed by the military as nerve gas agents, related comp.
such as parathion are employed as insecticides.
4 organophosphates are used in medicine these are Isoflurophate (DFP) and
echothiophate (are used as antiglaucoma agents). Malathion as a *scabicide and
metrifonate as * anthelmintic agent.
Toxicity of Irreversible Anti ChE:
This usually occurs because of accidental exposure to toxic amount of pesticides, the most
toxic of these drugs (e.g. parathion) are rapidly fetal if the exposure is not immediately
recognized and treated.
Signs and symptoms of poisoning: include general cholinergic stimulation (muscarinic
and nicotinic), paralysis of motor function "causing breathing difficulty," and CNS
stimulation leading to convulsions followed by respiratory and cardiovascular depression.

14
The spectrum of toxicity can be remembered with the aid of the mnemonic "DUMBELS"
D: diarrhea
U: urination
M: miosis (contraction of pupil)
B: bronchoconstriction
E: excitation of skeletal muscles and CNS followed by depression
L: lacrimation
S: salivation and sweating
Treatment of toxicity:
1-Contaminated clothing should be removed and the skin washed. Gastric lavage may
be needed if many of the substances have been ingested. Mechanical ventilation
may be needed
2-Atropine is the main stay of treatment; 2 mg is given IV or IM as soon as possible
and repeated every 15-60 minutes. A 100 mg of Atropine may be required for a
poisoned patient. Atropine antagonizes the muscarinic actions but has no effect
on the nicotinic signs of toxicity.
3-Enzyme reactivation (enzyme regeneration) by using enzyme regenerators,
compounds such as Pralidoxime are used, which may also reverse the nicotinic
signs if used early on "before aging occurs"
Aging:
Is a further chemical change in the enzyme which renders regenerator drugs unable to
remove the inhibitor (organophosphates phosphorylated the active site of the enzyme
cholinesterase, and following this covalent modification the phosphorylated enzyme
undergoes a further chemical change (losses an alkyl group) which is called aging that
makes it impossible for Pralidoxime to break the bond between the remaining
organophosphates and the enzyme.)
15
Pralidoxime (PAM):
Is an oxime compound. The oxime group has an extremely high affinity for the phosphate
atom in the organophosphates insecticides because the affinity of the oxime group for the
phosphorus exceeds that of the enzymes active site. This agent is therefore able to bind the
inhibitor and displace (regenerate) the enzyme if aging has not occurred. Pralidoxime is
given in a dose of 1 gm every 4 hours IM or by slow IV infusion.
Best results are obtained if it is given is within the first12 hours of poisoning.
Muscle power may improve within 30 min.
Cholinergic Antagonists
"cholinoceptor blockers"
Anti Muscarinic Anti Nicotinic
M1 selective Nonselective Ganglion blockers N-M blockers
Antimuscarinic drugs which act principally at postganglionic cholinergic
(parasympathetic) nerve endings, i.e. atropine-related drugs. Muscarinic receptors can
be subdivided according to their principal sites, namely in the brain and gastric parietal
cells (M1), heart (M2) and glandular and smooth muscle cells (M3).
Muscarinic Antagonists:
These agents (e.g. Atropine & Scopolamine) act as competitive "surmountable"
pharmacological antagonists.
They block muscarinic receptors causing inhibition of all muscarinic functions and
their blocking effects are overcome by increasing the concentration of Ach or by other
muscarinic agonists.
These drugs also block the few exceptional sympathetic neurons that are cholinergic
"e.g. sweat gland".
16
These drugs have little or no effect on the autonomic ganglia or the N-M junction.
Most of these drugs are nonselective for muscarinic receptors except for Pirenzepine
and telenzepine which are M1 selective (used in treatment of peptic ulcers).
The action of muscarinic blockers (e.g. atropine) is mostly predictable effects derived
from cholinoceptor blockade.
Effects of muscarinic blocking drugs:
Organs
Effect
Mechanism
CNS
Sedation, anti motion sickness action,
anti parkinsonian action, amnesia, delirium.
Block of MRs, unknown subtype.
Eye
Mydriasis, cycloplegia (paralysis of accommodation)
Block of M3 Rs
Bronchi
Bronchodilatation esp. if constricted.
Block of M3 Rs
GIT
Relaxation, slowed peristalsis.
Block of M1,M3 Rs
Genito-urinary tract
Relaxation of the bladder wall,
Urinary retention.
Block of M3 Rs
Heart
Initial bradychardia, esp. at low doses
then tachycardia.
Tachycardia from block of M2 R
in the heart.
Blood vessels
Block of muscarinic vasodilatation not manifested
unless a muscarinic agonist is present.
Block of M3 Rs on endothelium
of B.Vs
Glands
Marked reduction of salivation,
Lacrimation,sweating, less reduction
of gastric secretion.
Block of M1, M3 receptors.
Skeletal m.
none

17
Notes:
CNS effects are less predictable. CVS effects at therapeutic doses include an initial
slowing of the heart rate caused by a central mechanism (medullary stimulation of the
cardiac inhibitory centre) or more likely by presynaptic vagal effects (blocking the
presynaptic M1R on vagal post ganglionic fibers that normally limit Ach release in the SA
node).this slowing is followed by tachycardia and decrease AV conduction time as it would
be predicted from peripheral vagal blockade (blocking of M2 receptors in the heart).
The effect on the blood vessels include blockade of the endothelial muscarinic receptors
that mediate vasodilatation and also vasodilatation produced by sympathetic cholinergic
nerve stimulation to the skeletal muscle vascular bed. However there is a little effect on
blood pressure of normal persons. At toxic doses atropine may cause cutaneous
vasodilatation in the upper part of the body, the mechanism is unknown.
Clinical uses of muscarinic antagonists:
Note:
Topical activity (the ability to enter the eye after conjuctival administration) and similar
ability to cross lipid barrier (e.g. BBB) is important in determining the usefulness of several
antimuscarinics often used in ophthalmology and in Parkinsonism. In contrast the
antimuscarinic drugs used for their antisecretory or antispasmodic actions in the gut or in the
bronchi are often selected for minimum CNS activity (these drugs may incorporate
quaternary amine group to limit penetration to the CNS).
1-CNS:
Motion sickness: scopolamine is the standard therapy in motion sickness. They are used as
Antiemetic (principally hyoscine, promethazine).Their sedative action is used in
anaesthetic
premedication
(hyoscine).Parkinsonism:
Benztropine,
biperidine,
trihexphenidyl, benzhexol (these drugs are mainly effective to control tremors).
Benztropine is sometimes used to treat acute dystonias caused by
Antipsychotic drugs.
18
2- Eye:
Are used to dilate the pupil (mydriasis) and to paralyze accommodation (cycloplegia).
Drugs used to this purpose include (in descending order of duration of action).
Atropine <72 hours,
homatropine (24 hrs.): shorter duration than atropine and less likely cause serious increase
in IOP. Complete cycloplegia cannot always be obtained unless repeated instillation of eye
drop (1% and 2%) is made every 15 min for 1-2 h.
cyclopentolate (2-12 hrs.), tropicamide (0.5-4 hrs.).
Tropicamide is the shortest acting of the mydriatics.
3-Bronchi:
Parentral atropine has been used to reduce airway secretion during surgery.
Ipratropium a quaternary agent is used by inhalation to reduce bronchoconstriction in
asthma and COPD (chronic obstructive pulmonary disease). This drug has very few
Antimuscarinic effects outside the lung because it is rapidly metabolized and poorly
absorbed. It is less effective than B2 agonists in this regard.
Tiotopium: is alternative to ipratropium but only for patient with COPD. It is not licensed
for acute bronchconstriction because of its slow oncet of action.
4- CVS:
Parentral atropine and similar antimuscarinics are used to H.R in cases of bradychardia
due to depression of the SA node or the AV node function (e.g. after myocardial infarction
(MI) and in persons with hyperactive carotid sinus reflexes).
Atropine is an important antagonist of both central nervous, parasympathomimetic and
vasodilator effects, though it has no effect at the neuromuscular junction and will not
prevent voluntary muscle paralysis.

19
5- Gut:
Atropine, methscopolamine and propantheline{ decrease smooth muscle relaxtion e.g
IBS (IRRITABLE BOWELL SYNDOME)} were used in the past in acid peptic disease
to reduce acid secretion (volume, amount of acid and pepsin and mucin are all reduced but
large doses are required).
These drugs are less effective than the H2 antagonist (e.g. cimitidine) and produce
frequent side effects.
Pirenzepine and telenzepine are M1, selective antagonist, may be more useful in
peptic ulcer (M1 receptors are present on the ECL cells and not on parietal cells
which possess M3R.)
Muscarinic antagonist e.g. glycopyrolate, dicyclomine, Hyoscine butyl bromide
and methscopolamine can be used to reduce cramping (antispasmodic) and
hypermotility in transient diarrheas but diphenoxylate (an Opioid) is more
effective, and to reduce morphine-induced smooth muscle spasm when the
analgesic is used against acute colic.
6- Urinary system:
Oxybutynin, glycopyrolate, dicyclomine, propiverine, trospium are used in the treatment
of spasm and urgency induced by mild inflammation e.g. (Cystitis), surgery and
neurological conditions but can precipitate urinary retention in elderly men with benign
prostatic hyperplasia (Bph).
Tolterodine (is an M3 selective antimuscarinics) is used in adult with urinary
incontinence.
Glycopyrolate:
preanaesthetic
medication
and
reduce
salivation
I.V
administration cause less tachycardia than atropine.
Dicyclomine: used in management of ZES
7- Other uses:
As an antidote for cholinergic agonists atropine is used in the treatment of Organophosphorus
poisoning and some types of mushroom poisoning. Atropine because of its ability to enter
CNS is used to block the effect of excess Ach resulting from inhibition of ChE by drugs such
as Physostigmine.

20
For their peripheral actions: In anaesthesic premedication, atropine, and Hyoscine* block
the vagus and reduce mucosal secretions; Hyoscine also has useful sedative effects.
Glycopyrronium* is frequently used during anaesthetic recovery to block the muscarinic
effects of neostigmine given to reverse a nondepolarising neuromuscular blockade.
In the respiratory tract, ipratropium* is a useful bronchodilator in chronic obstructive
pulmonary disease and acute asthma.
Atropine:
Atropine is the prototype drug of this group and will be described first. Other named agents
will be mentioned only in so far as they differ from atropine. All act as non-selective and
competitive antagonists of the various muscarinic receptor subtypes (Ml-3).It is an alkaloid
found in atropa belladonna. It is a tertiary amine (lipid soluble and crosses to the CNS). It is
eliminated partly by metabolism and by renal excretion t ½ is 2 hrs. Duration of action is 4-8
hrs except in the eye where the effect lasts for 72 hours or longer. The antidote in atropine
poisoning is Physostigmine.
Scopolamine (Hyoscine):
Is another belladonna alkaloid similar to atropine but has a longer duration of action and
greater action on the CNS. It produces sedation. Its use is limited for the prevention and
treatment of motion sickness.
Adverse effects and toxicity: Most of the adverse effects are predictable Antimuscarinic
effects, although there are some unpredictable actions.
Predictable include: Dry mouth, blurred vision, tachycardia, constipation,
hyperthermia due to blockage of thermo-regulatory sweating also called atropine fever
(this occurs esp. in infants which may be dangerous.)
Atropine toxicity is described as feeling as dry as a bone because sweating, salivation
and lacrimation are reduced or stopped. In the elderly atropine may cause acute angle-
closure glaucoma and urinary retention esp. in patients with Bph (benign prostatic
hyperplasia)

21
Unpredictable include:
1- CNS toxicity which includes: sedation, amnesia and delirium or hallucination
(described as mad as a hatter) convulsions may also occur.
2- CVS which include: intra ventricular conduction block and cutaneous
vasodilatation of the vessels of head, neck, arms and trunk (atropine flush) which is
described as red as beet, which may be diagnostic.
Contra indications:
These drugs should be cautiously used in infants (hyperthermia) these drugs are relatively CI
in patients with glaucoma and in men with Bph.
Nicotinic antagonist:
A-Ganglionic blocking drugs:
These agents act specifically on nicotinic receptors by blocking the ion channels in the
ganglia both sympathetic and parasympathetic. Thus they block the entire output of the
ANS at the nicotinic receptors. They have no effect on N-M junction .they were the 1
st
successful agents in the treatment of hypertension. However because of their adverse effect,
they are rarely used nowadays. Examples of these drugs are hexamothanium,
mecamylamine and trimetaphan.
Nicotine: present in tobacco smoke and is used in the form of chewing gum or
transdermal patches by smokers to get rid of smoking and is still used in some
insecticides. Depending on the dose nicotine 1
st
stimulate the ganglia which are
followed by blockade. The stimulatory effects are complex which include CNS
stimulation Bp, HR (due to increase transmitter release from adrenergic nerve
terminals and adrenal medulla), peristalsis and secretion and ADH release.
At higher doses Bp because of Ganglionic blockade and the activity of the smooth
muscles and secretion is blocked.
Mecamylamine
Mecamylamine produces a competitive nicotinic blockade of the ganglia.
Mecamylamine has been supplanted by superior agents with fewer side effects.
USED FOR treatment of moderately sever and sever hypertension.

22
B- Neuromuscular blocking drugs:
These neuromuscular blockers are structural analogues of Ach they are either of:
1- A non- depolarizing type: (act as antagonists) e.g. tubocurarine.
2- A depolarizing type (act as agonists) e.g. succinyl choline.
N-M blockers are clinically useful during surgery to produce complete muscle relaxation
without the need to employ high anesthetic doses to achieve a comparable muscle relaxation.
Non-depolarizing type (competitive N-M blockers):
(Tubocurarine, pancuronium, gallamine, Rocuronium, atracurium, mivacurium,
vecuronium, doxacurium).
All of these drugs are given parentraly. They differ in their onset and duration of action
(Rocuronium has the fastest onset of action), whether they are metabolized by plasma
choline esterase (e.g. mivacurium) or eliminated in bile (e.g.vecuronium) or by the kidney
(e.g. doxacurium, pancuronium, tubocurarine) or eliminated by an independent mechanism
(e.g. atracurium) which involves spontaneous breakdown called (Hoffmann elimination).
Also these drugs differ in their autonomic effects and their ability to release histamine e.g.
tubocurarine blocks the ganglia and is the most likely of N-M blockers to cause histamine
release. Histamine release also occurs to a less extent with atracurium and mivacurium, on
the other hand pancuronium and gallamine block the cardiac muscarinic receptors causing
tachycardia.
Mechanism of action of non-depolarizing agents:
These drugs compete with Ach and produce a competitive block at the end plate nicotinic
receptors and thus prevent the depolarization of the muscle cell membrane causing flaccid
paralysis.
The action of these drugs can be overcome by the concentration of Ach at the synaptic
cleft e.g. by administration of ChE inhibitors (Neostigmine or Edrophonium) some drugs
in this group or when given in high doses may directly act to plug (close) the ion channels of
the end plate, this leads to further weakening of N-M transmission and reduces the ability of
ChE inhibitors to reverse the action of these drugs.

23
A. At low doses: Nondepolarizing neuromuscular-blocking drugs interact with the nicotinic
receptors to prevent the binding of ACh (Figure 5.9). Thus, these drugs prevent
depolarization of the muscle cell membrane and inhibit muscular contraction. Because these
agents compete with ACh at the receptor without stimulating it, they are called competitive
blockers. Their action can be overcome by increasing the concentration of ACh in the
synaptic gap, for example, by administration of such cholinesterase inhibitors as
neostigmine, pyridostigmine, and edrophonium. Anesthesiologists often employ this strategy
to shorten the duration of the neuromuscular blockade.
B. At high doses: Nondepolarizing blockers can block the ion channels of the endplate. This
leads to further weakening of neuromuscular transmission, thereby reducing the ability of
AChE inhibitors to reverse the actions of the non depolarizing muscle relaxants. With
complete blockade, no direct electrical stimulation is seen.
Actions:
Small rapidly contracting muscles of the face and eye are the most susceptible to blockage
and are paralyzed first followed by the muscles of the fingers then the limbs then neck and
trunk muscles then the intercostals muscles and lastly the diaphragm. The muscles recover in
the reverse manner, with the diaphragm muscles recovering first and contracting muscles of
the face and the eye recovering last. Those agents that release histamine (for example,
atracurium) can produce a fall in blood pressure, flushing, and bronchoconstriction.
Therapeutic uses:
These drugs are used mainly as surgical adjuvant to anesthesia for promoting skeletal
muscle relaxation and for facilitating endotracheal intubation.
Pharmacokinetics:
All neuromuscular-blocking agents are injected intravenously because their uptake via oral
absorption is minimal. These agents possess two or more quaternary amines in their bulky
ring structure, making them orally ineffective. They penetrate membranes very poorly and
do not enter cells or cross the blood-brain barrier.

24
Drug interactions:
a. Cholinesterase inhibitors:
Drugs such as neostigmine, physostigmine, pyridostigmine, and edrophonium can overcome
the action of nondepolarizing neuromuscular blockers, but, with increased dosage,
cholinesterase inhibitors can cause a depolarizing block as a result of elevated ACh
concentrations at the endplate membrane. If the neuromuscular blocker has entered the ion
channel, cholinesterase inhibitors are not as effective in overcoming blockade.
b. Halogenated hydrocarbon anesthetics:
Drugs such as halothane act to enhance neuromuscular blockade by exerting a stabilizing
action at the NMJ. These agents sensitize the NMJ to the effects of neuromuscular blockers.
c. Aminoglycoside antibiotics:
Drugs such as gentamicin and tobramycin inhibit ACh release from cholinergic nerves by
competing with calcium ions. They synergize with pancuronium and other competitive
blockers, enhancing the blockade.
d. Calcium-channel blockers:
These agents may increase the neuromuscular block of competitive blockers as well as
depolarizing blockers.
Adverse effects:
1- Respiratory paralysis if mechanical ventelation is not provided.
2- Autonomic effect and histamine release depending on the drugs used (e.g. tubocurarine.
Drug interactions:
Halogenated hydrocarbon anesthetics (halothane, Enflurane), aminoglycoside antibiotics
(e.g. gentamycin, tobramycin), calcium channel blockers and some antiarrythmic drugs
(e.g. quinidine) increase the effect of N-M blockers. On the other hand ChE inhibitors
antagonize their effect.

25
Depolarizing agents:
Succinylcholine (suxamethonium), decamethonium
Mechanism of action
These depolarizing agents act like Ach on the nicotinic receptors of the motor end plate to
depolarize the junction, but unlike Ach, Succinylcholine (because is not rapidly hydrolyzed
by ChE) remains attached to the receptor for a relatively longer time providing a constant
stimulation of the receptor.
Succinylcholine therefore first causes the opening of Na – channel associated with nicotinic
receptor which results in depolarization of the receptor (phase I) this leads to a transient
twitching of the muscle fasciculation (involuntary muscle contraction and relaxation) then
flaccid paralysis.
The continued binding of the depolarizing agent render the receptor incapable of
transmitting further impulses (desensitization) and results in flaccid paralysis (phase II) in
which the membrane depolarizes but the receptor is desensitized to the effect of Ach.(a
curare like effect). If a cholinesterase inhibitor is given, phase I is augmented but it may
reverse phase II block.
Therapeutic uses:
Because of its rapid onset and short duration of action (only few minutes if given as single
dose) succinyl choline is useful for endotracheal intubation during induction of anesthesia
and may be also used during ECT (electro-convulsive therapy).
Because of its brief duration of action (due to its rapid hydrolysis by plasma ChE) it is
usually given by a continuous infusion.
26
Adverse effects:
Malignant hyperthermia in genetically susceptible individual when succinyl choline is
used in combination with Halothane (anesthetic), hyperpyrexia and muscle rigidity may
occur, this is treated by rapidly cooling the body and by administration of Dantroline which
blocks the release of Ca from sarcoplasmic reticulum of the muscle cell thus reducing heat
and decreasing muscle rigidity which is associated with hyperpyrexia.
Apnea: a genetically related deficiency of plasma ChE or the presence of atypical form of
the enzyme can lead to prolonged apnea.
Other adverse effects include: post operative muscle pain, intragastric pressure which
may promote emesis (vomiting), IOP, hyperkalemia (especially in patients with burns).
Succinyl choline may cause stimulation of the autonomic ganglia and cardiac muscarinic
receptor (bradychardia) and has a slight ability to release histamine.