
Dr.Ahmed Rushdi

Drugs have been used for the treatment of infectious
diseases since the 17th century (eg, quinine for
malaria, emetine for amebiasis).
however,
chemotherapy as a science began in the
first decade of the 20th century with understanding
of the principles of selective toxicity
, the specific
chemical relationships between microbial pathogens
and drugs, the development of drug resistance, and
the role of combined therapy.
Experiments led to the arsphenamines for syphilis,
the first planned chemotherapeutic regimen.

The current era of antimicrobial chemotherapy
began in 1935 with the discovery of the
sulfonamides.
In 1940, it was demonstrated that penicillin,
discovered in 1929, could be an effective therapeutic
substance.
During the next 25 years, research on
chemotherapeutic agents centered largely around
substances of microbial origin called antibiotics.
The isolation, concentration, purification, and mass
production of penicillin were followed by the
development
of
streptomycin,
tetracyclines,
chloramphenicol, and many other agents.

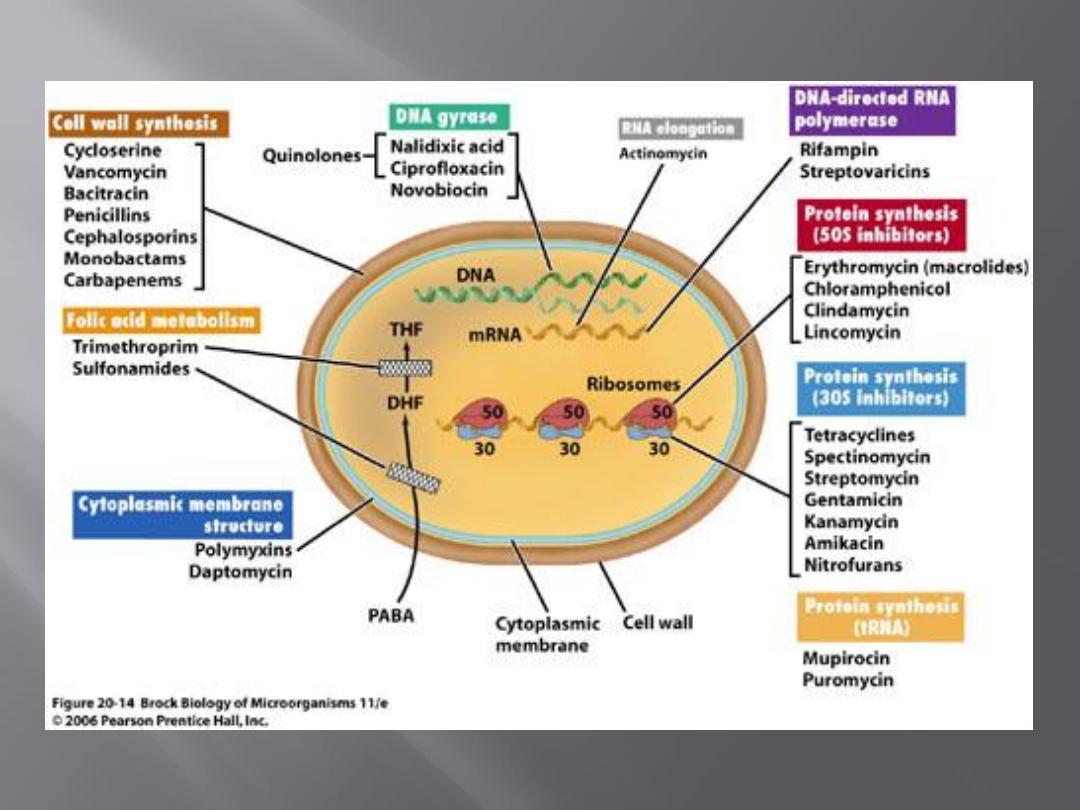
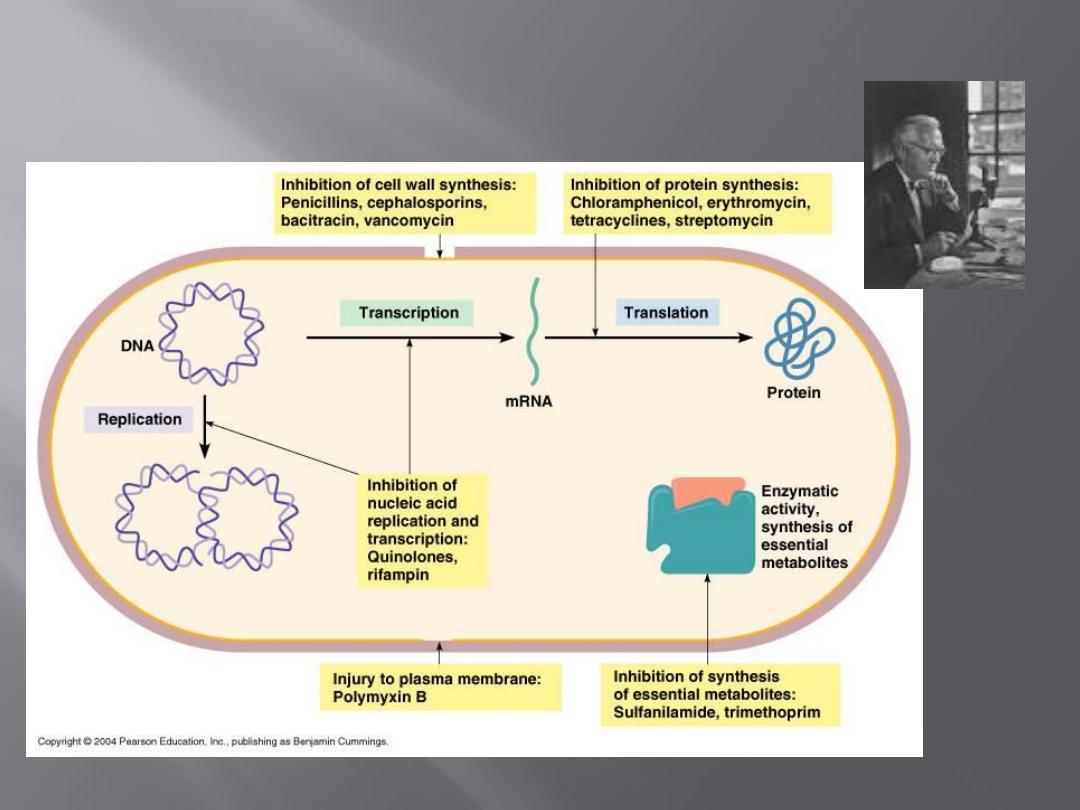
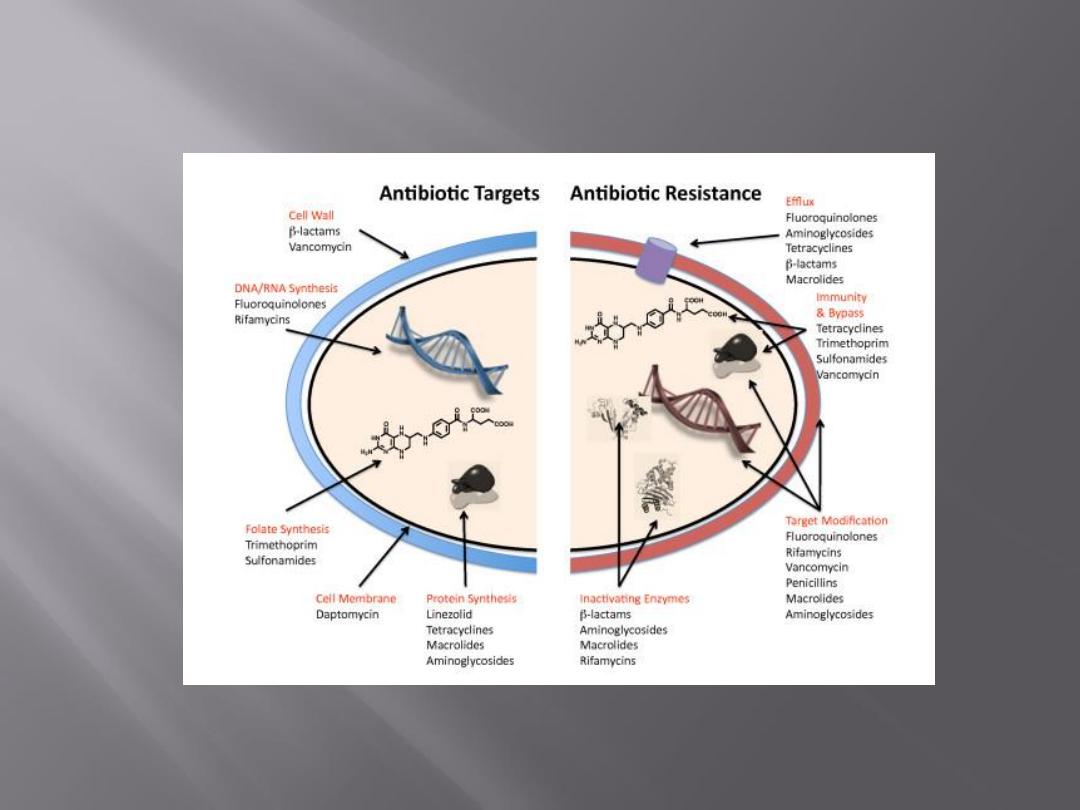
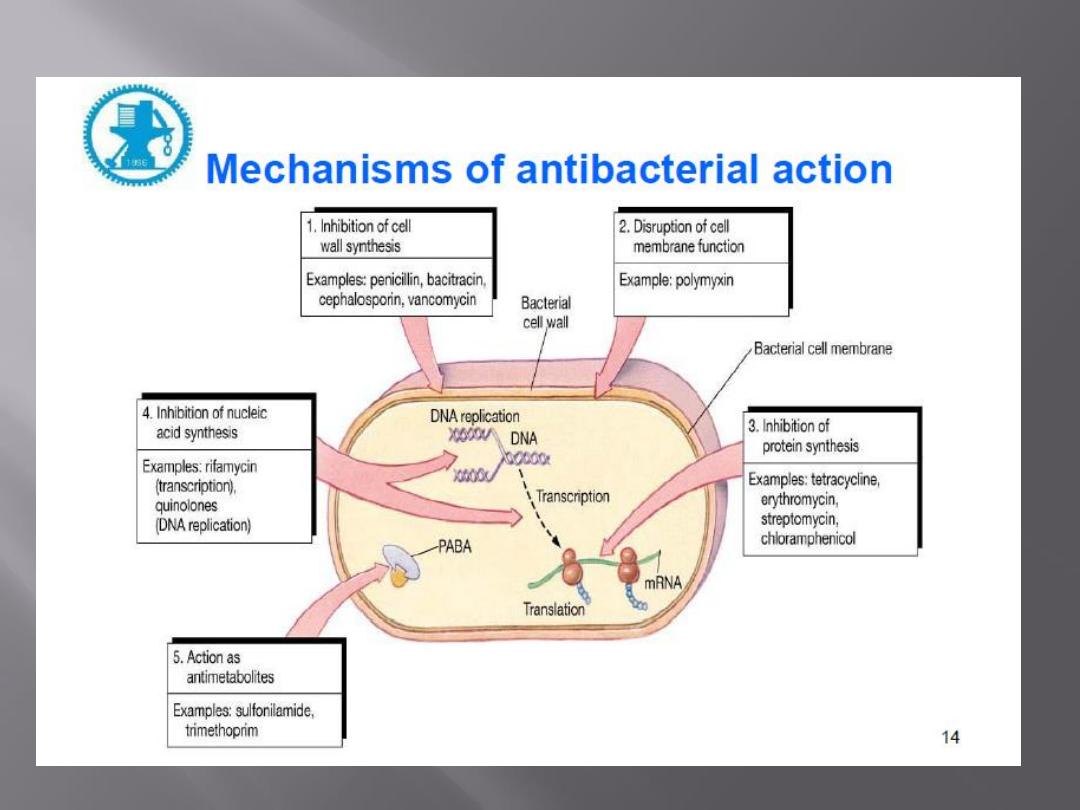
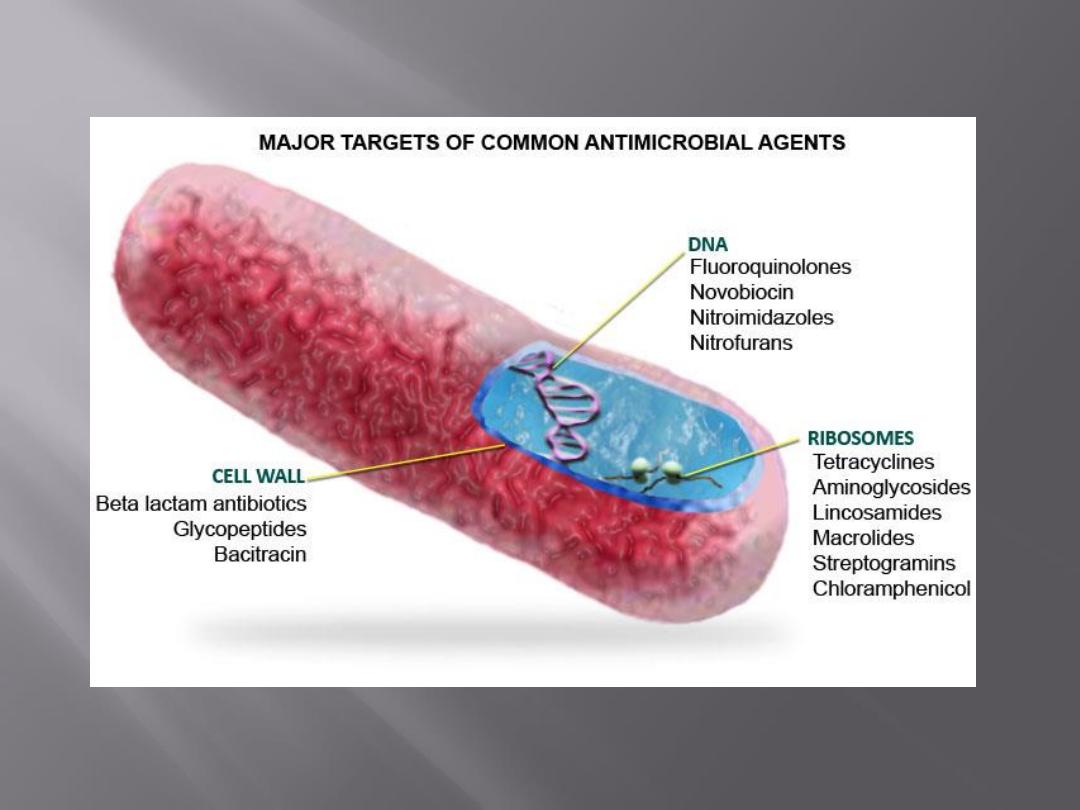
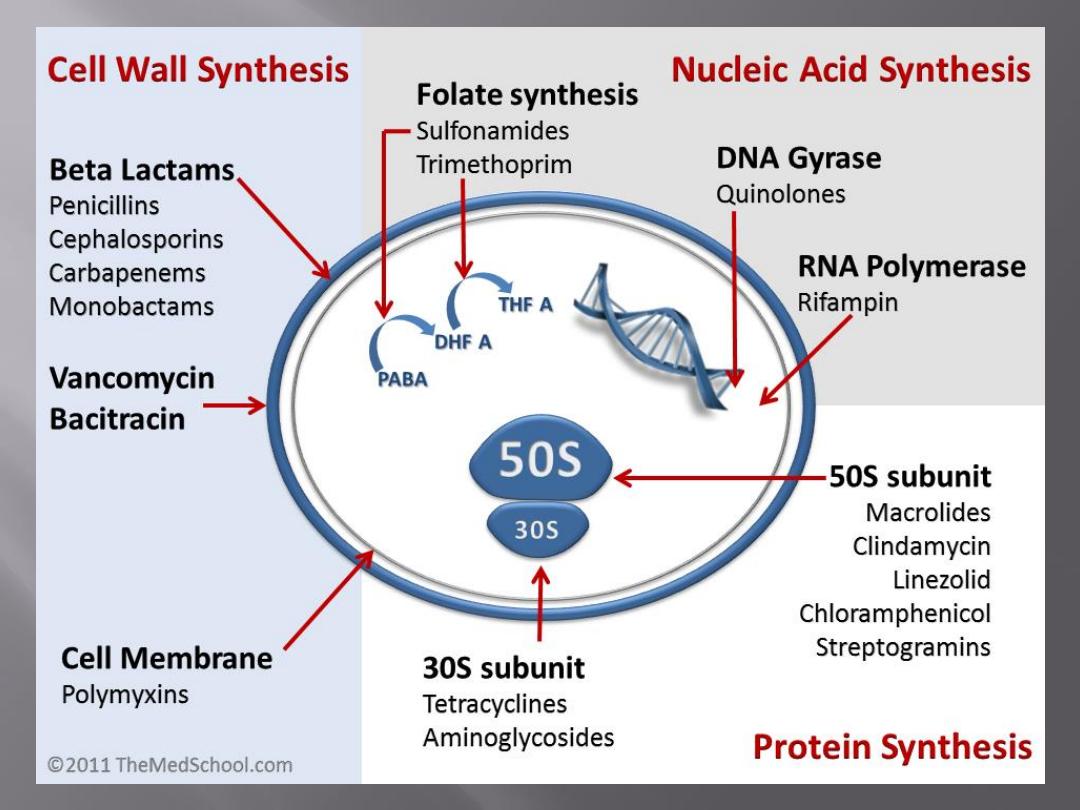
Antimicrobial drugs act in one of several
ways:
by selective toxicity,
by inhibition of cell membrane synthesis
and function,
by inhibition of protein synthesis, or
by inhibition of nucleic acid synthesis

An ideal antimicrobial agent exhibits selective
toxicity, which means that the drug is harmful to a
pathogen without being harmful to the host.
Often, selective toxicity is relative rather than
absolute; this implies that a drug in a concentration
tolerated by the host may damage an infecting
microorganism.
Selective toxicity may be a function of a specific
receptor required for drug attachment, or it may
depend on the inhibition of biochemical events
essential to the pathogen but not to the host.

Bacteria have a rigid outer layer, the cell wall. The cell wall
maintains the shape and size of the microorganism, which
has a high internal osmotic pressure. Injury to the cell wall
(eg, by lysozyme) or inhibition of its formation may lead to
lysis of the cell.
In a hypertonic environment (eg, 20% sucrose), damaged cell
wall formation leads to formation of spherical bacterial
"protoplasts"
from
gram-positive
organisms
or
"spheroplasts" from gram-negative organisms;
these forms are limited by the fragile cytoplasmic membrane.
If such protoplasts or spheroplasts are placed in an
environment of ordinary tonicity, they take up fluid rapidly,
swell, and may explode.
Specimens from patients being treated with cell wall-active
antibiotics often show swollen or misshapen bacteria.
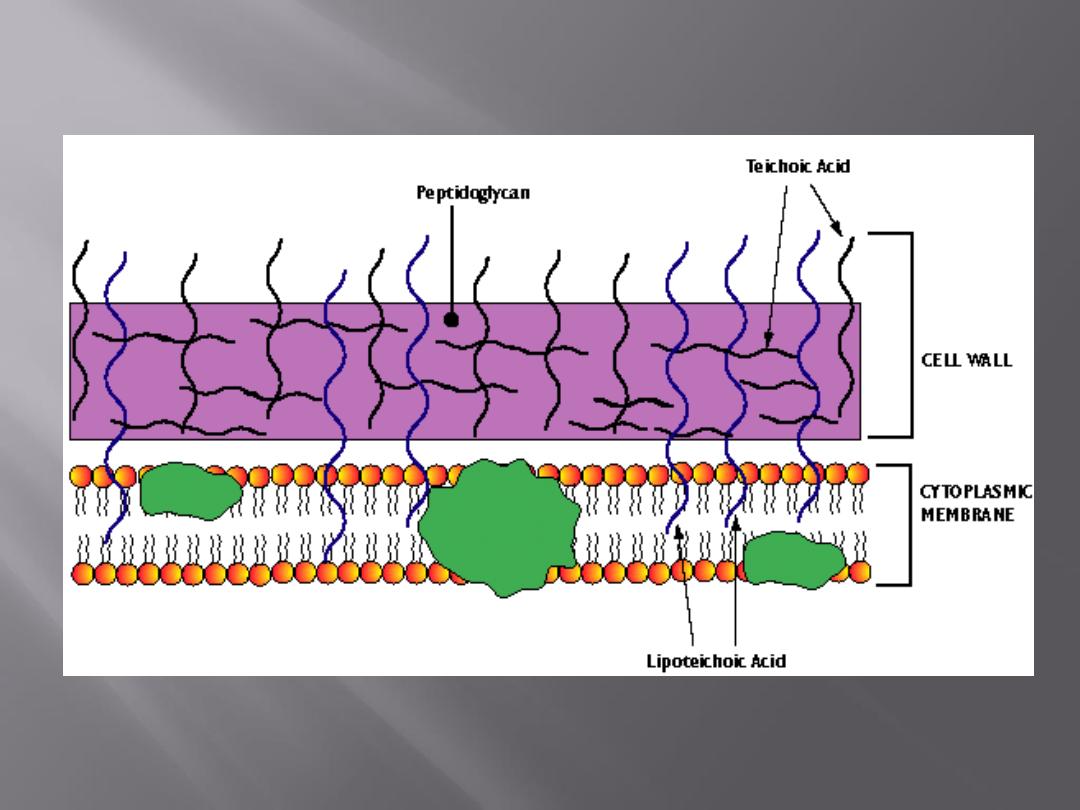

The cell wall contains a chemically distinct complex
polymer
"mucopeptide"
("peptidoglycan")
consisting of polysaccharides and a highly cross-
linked polypeptide.
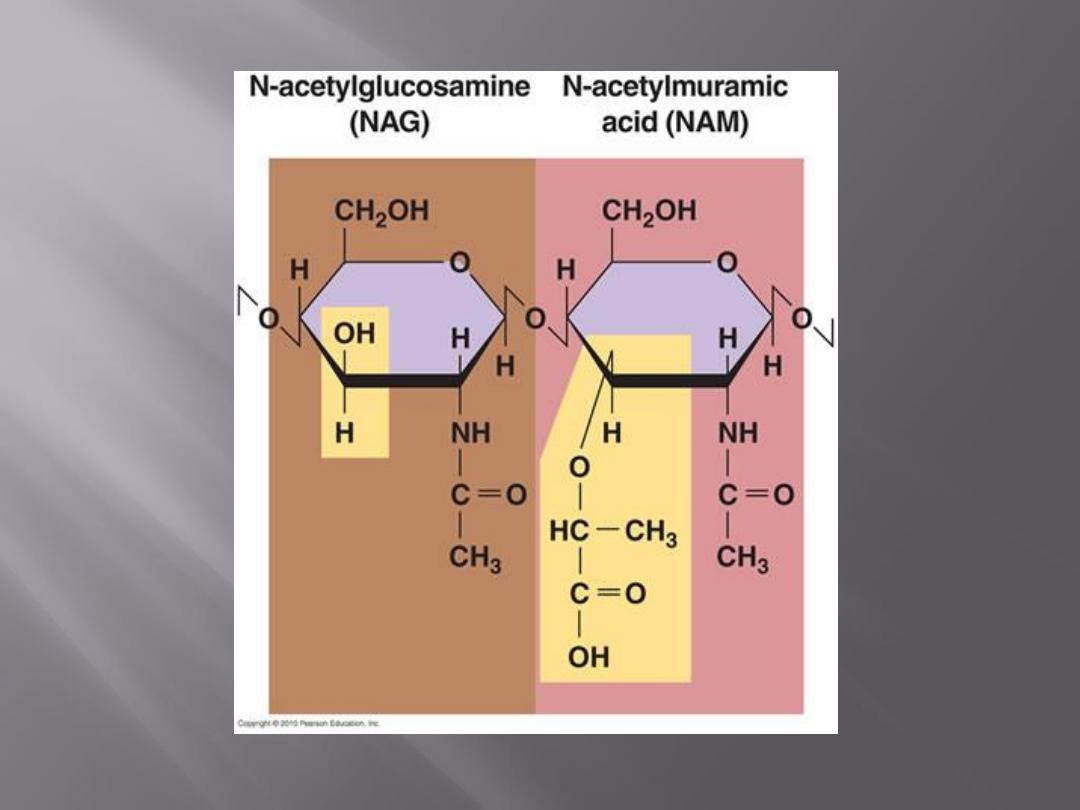
The polysaccharides regularly contain the amino
sugars N-acetylglucosamine and acetylmuramic
acid. The latter is found only in bacteria. To the
amino sugars are attached short peptide chains.
The peptidoglycan layer is much thicker in the cell
wall of gram-positive than of gram-negative
bacteria.

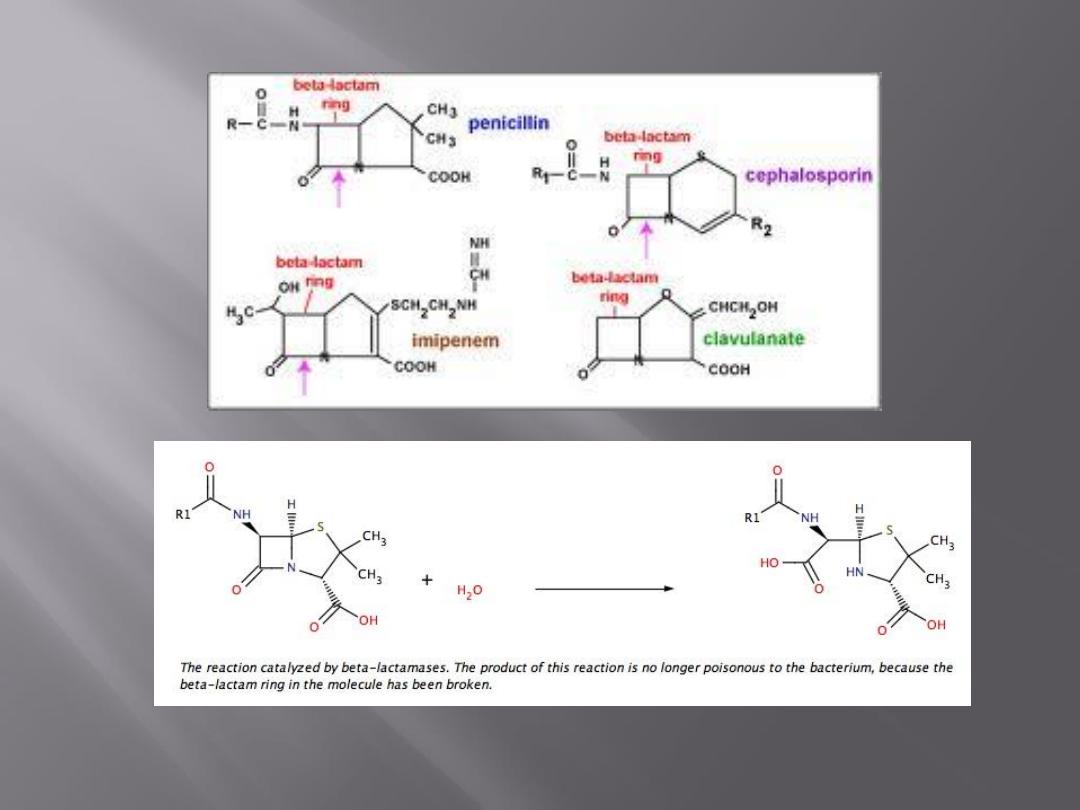
All -lactam drugs are selective inhibitors of bacterial
cell wall synthesis and therefore active against
growing bacteria.
This inhibition is only one of several different
activities of these drugs, but it is the best
understood.
The initial step in drug action consists of binding of
the drug to cell receptors (penicillin-binding
proteins; PBPs).
There are three to six PBPs (MW 4–12 x 105), some
of which are transpeptidation enzymes.
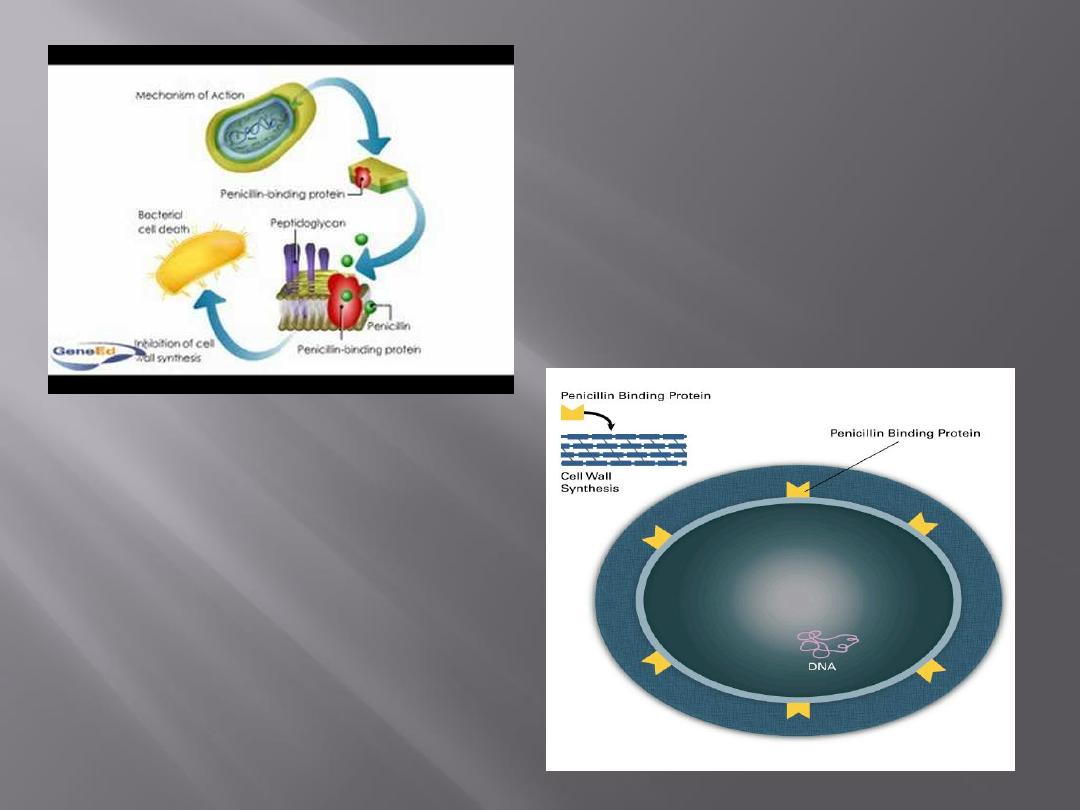

Different receptors have different affinities for a
drug, and each may mediate a different effect.
For example, attachment of penicillin to one PBP
may result chiefly in abnormal elongation of the cell,
whereas attachment to another PBP may lead to a
defect in the periphery of the cell wall, with resulting
cell lysis
.
PBPs are under chromosomal control, and mutations
may alter their number or their affinity for -lactam
drugs.

Resistance to penicillins may be determined by the
organism's production of penicillin-destroying enzymes (-
lactamases). -Lactamases open the -lactam ring of penicillins
and cephalosporins and abolish their antimicrobial activity.
-Lactamases have been described for many species of gram-
positive and gram-negative bacteria. Some -lactamases are
plasmid-mediated (eg, penicillinase of S aureus), while
others are chromosomally mediated (eg, many species of
gram-negative bacteria).
All of the more than 30 plasmid-mediated -lactamases are
produced constitutively and have a high propensity to move
from one species of bacteria to another (eg, -lactamase-
producing Neisseria gonorrhoeae, Haemophilus influenzae, and
enterococci).
Chromosomally mediated -lactamases may be constitutively
produced (eg, Bacteroides, Acinetobacter), or they may be
inducible (eg, Enterobacter, Citrobacter, Pseudomonas).

The cytoplasm of all living cells is bounded by the
cytoplasmic membrane, which serves as a selective
permeability barrier, carries out active transport
functions, and thus controls the internal composition of
the cell.
If the functional integrity of the cytoplasmic membrane
is disrupted, macromolecules and ions escape from the
cell, and cell damage or death ensues.
The cytoplasmic membrane of bacteria and fungi has a
structure different from that of animal cells and can be
more
readily
disrupted
by
certain
agents.
Consequently, selective chemotherapy is possible.
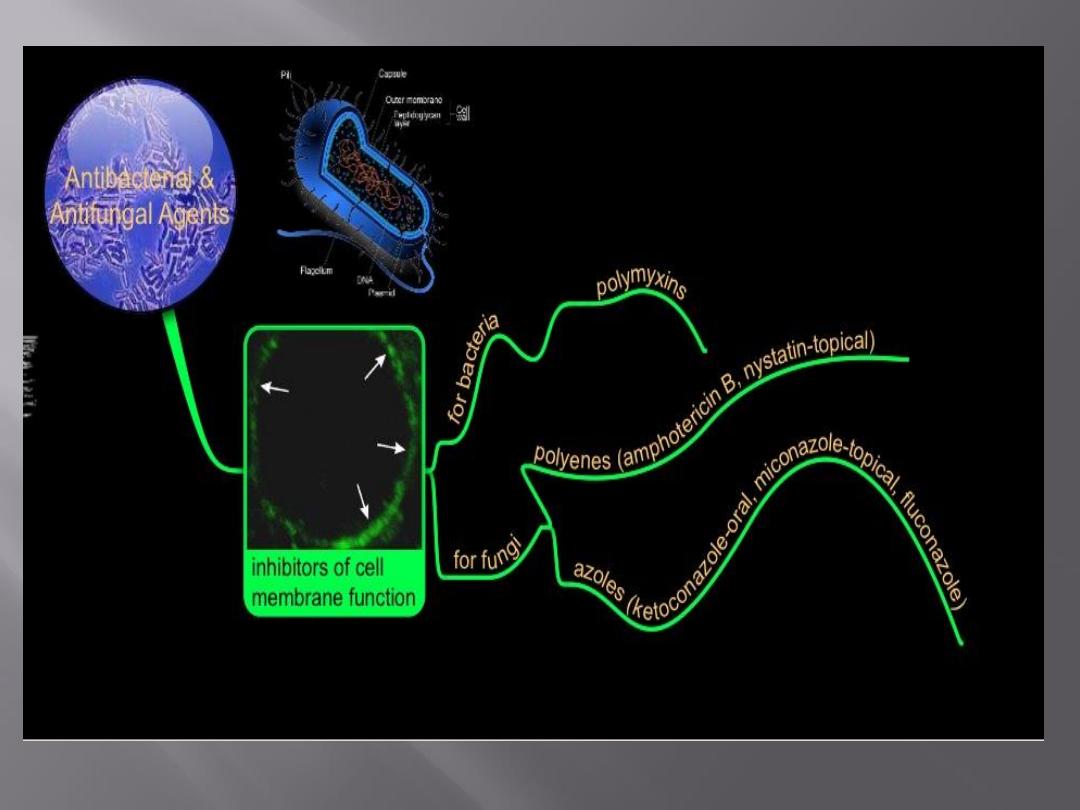

Detergents, which contain lipophilic and hydrophilic
groups, disrupt cytoplasmic membranes and kill the
cell .
One class of antibiotics, the
polymyxins
, consists of
detergent-like cyclic peptides that selectively damage
membranes containing phosphatidylethanolamine, a
major component of bacterial membranes.
A number of antibiotics specifically interfere with
biosynthetic functions of the cytoplasmic membranes—
eg, nalidixic acid and novobiocin inhibit DNA
synthesis, and novobiocin also inhibits teichoic acid
synthesis.


A third class of membrane-active agents is the
ionophores, compounds that permit rapid diffusion of
specific cations through the membrane.
Valinomycin, for example, specifically mediates the
passage of potassium ions.
Some ionophores act by forming hydrophilic pores in
the membrane; others act as lipid-soluble ion carriers
that behave as though they shuttle back and forth
within the membrane.
Ionophores can kill cells by discharging the membrane
potential,
which
is
essential
for
oxidative
phosphorylation, as well as for other membrane-
mediated processes; they are not selective for bacteria
but act on the membranes of all cells.

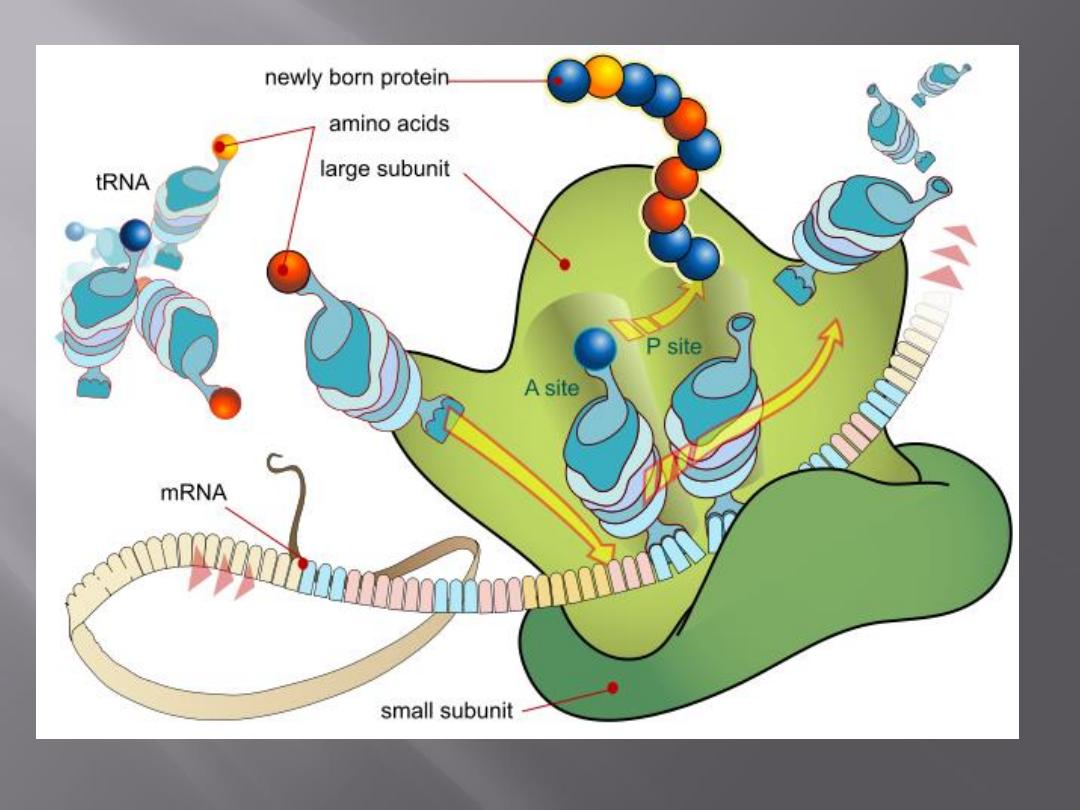
Bacteria have 70S ribosomes, whereas mammalian
cells have 80S ribosomes.
In normal microbial protein synthesis, the mRNA
message is simultaneously "read" by several
ribosomes that are strung out along the mRNA
strand. These are called
polysomes.
Examples of drugs acting by inhibition of protein
synthesis are the
erythromycins, lincomycins,
tetracyclines, glycylcyclines, aminoglycosides, and
chloramphenicol.

The mode of action of streptomycin has been studied far more
intensively than that of other aminoglycosides, but all probably act
similarly.
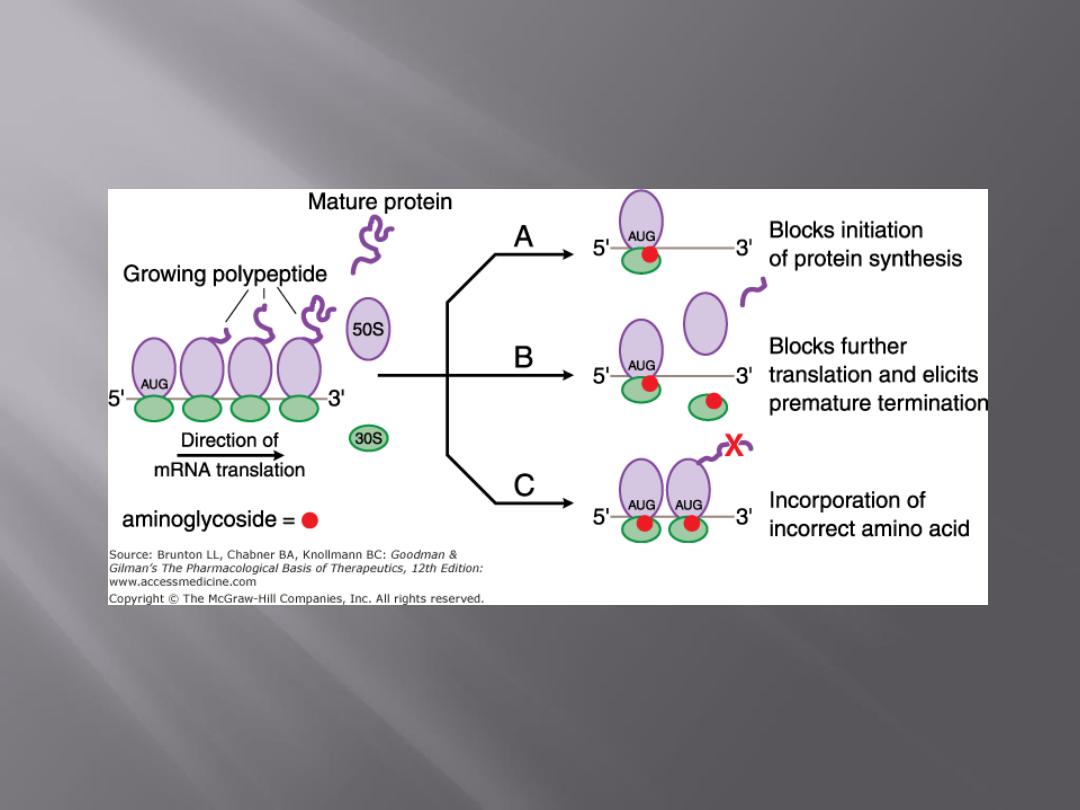
The first step
is the attachment of the aminoglycoside to a specific
receptor protein (P 12 in the case of streptomycin) on the 30S
subunit of the microbial ribosome.
Second,
the aminoglycoside blocks the normal activity of the
"initiation complex" of peptide formation (mRNA + formyl
methionine + tRNA).
Third,
the mRNA message is misread on the "recognition region"
of the ribosome; consequently, the wrong amino acid is inserted
into the peptide, resulting in a nonfunctional protein.
Fourth,
aminoglycoside attachment results in the breakup of
polysomes and their separation into monosomes incapable of
protein synthesis.
These activities occur more or less simultaneously, and the overall
effect is usually an irreversible event—killing of the bacterium.


Chromosomal
resistance
of
microbes
to
aminoglycosides principally depends on the lack of
a specific protein receptor on the 30S subunit of the
ribosome.
Plasmid-dependent resistance
to aminoglycosides
depends on the production by the microorganism of
adenylylating, phosphorylating, or acetylating
enzymes that destroy the drugs.
A third type of resistance consists of a
"permeability
defect,"
an outer membrane change that reduces
active transport of the aminoglycoside into the cell
so that the drug cannot reach the ribosome. Often
this is plasmid-mediated.

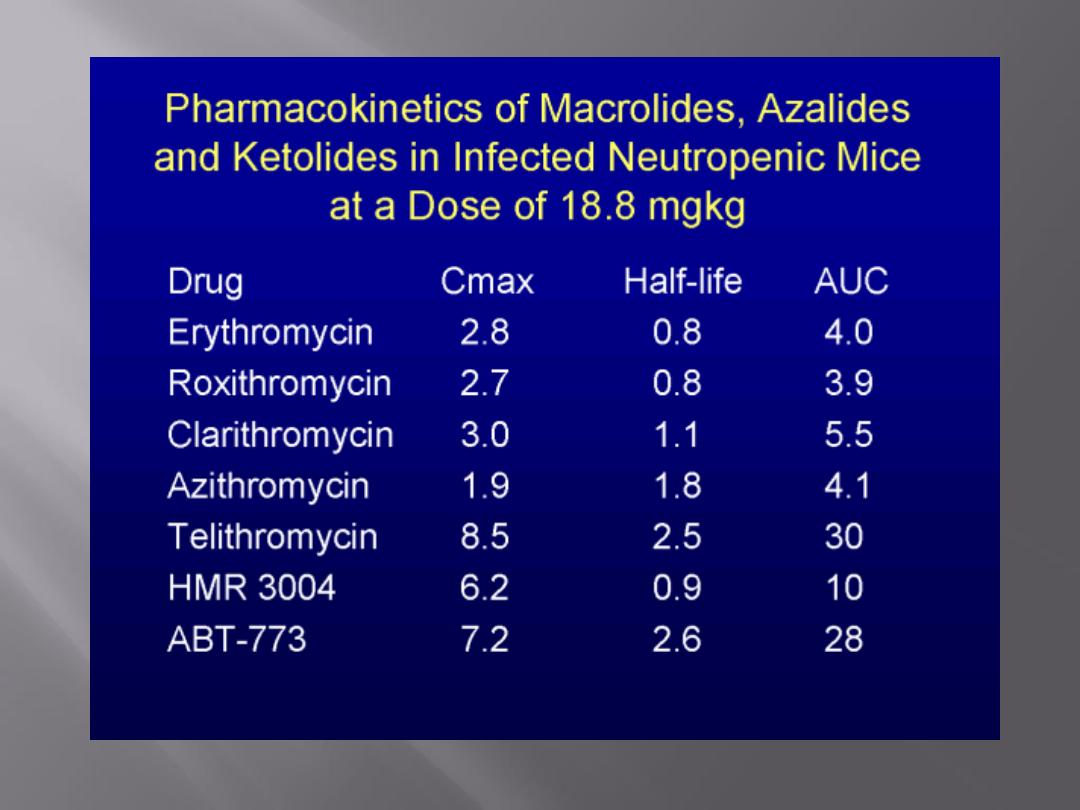
These
drugs
(erythromycins,
azithromycin,
clarithromycin, and roxithromycin and the ketolide,
telithromycin) bind to the 50S subunit of the
ribosome, and the binding site is a 23S rRNA.
They may interfere with formation of initiation
complexes for peptide chain synthesis or may
interfere with aminoacyl translocation reactions.
Some macrolide-resistant bacteria lack the proper
receptor on the ribosome (through methylation of
the rRNA). This may be under plasmid or
chromosomal control.

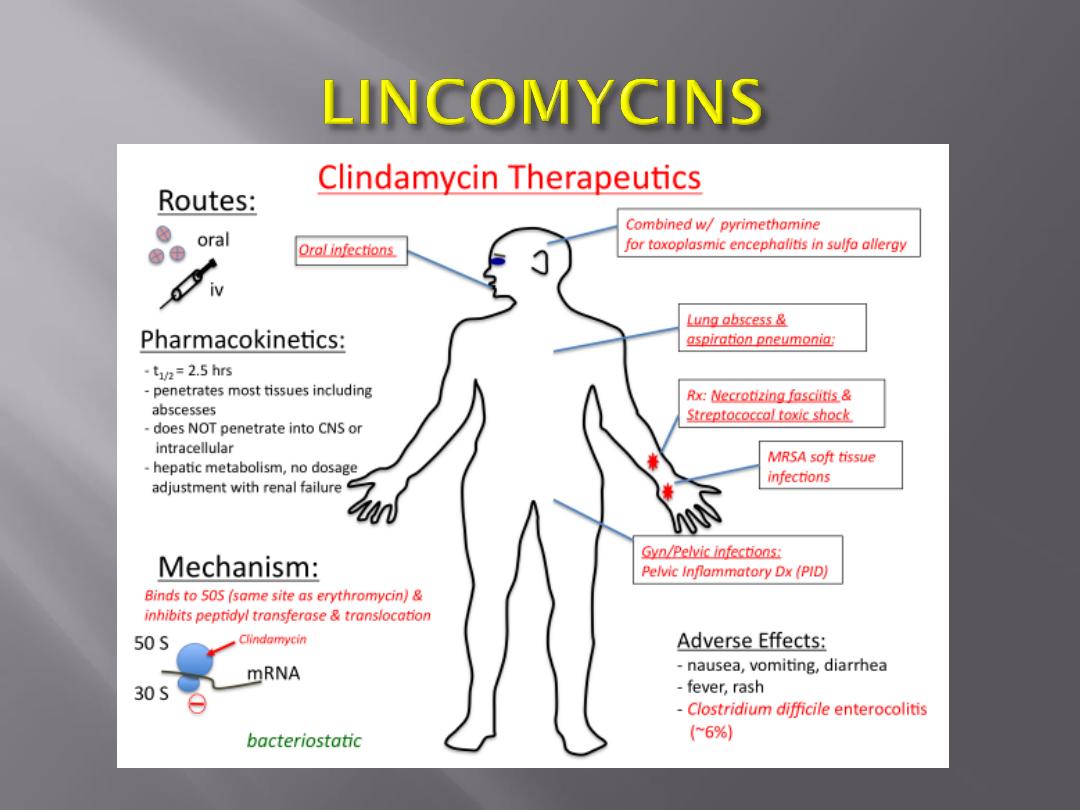
Clindamycin binds to the 50S subunit of the
microbial ribosome and resembles macrolides in
binding site, antibacterial activity, and mode of
action.
Chromosomal mutants are resistant because
they lack the proper binding site on the 50S
subunit.

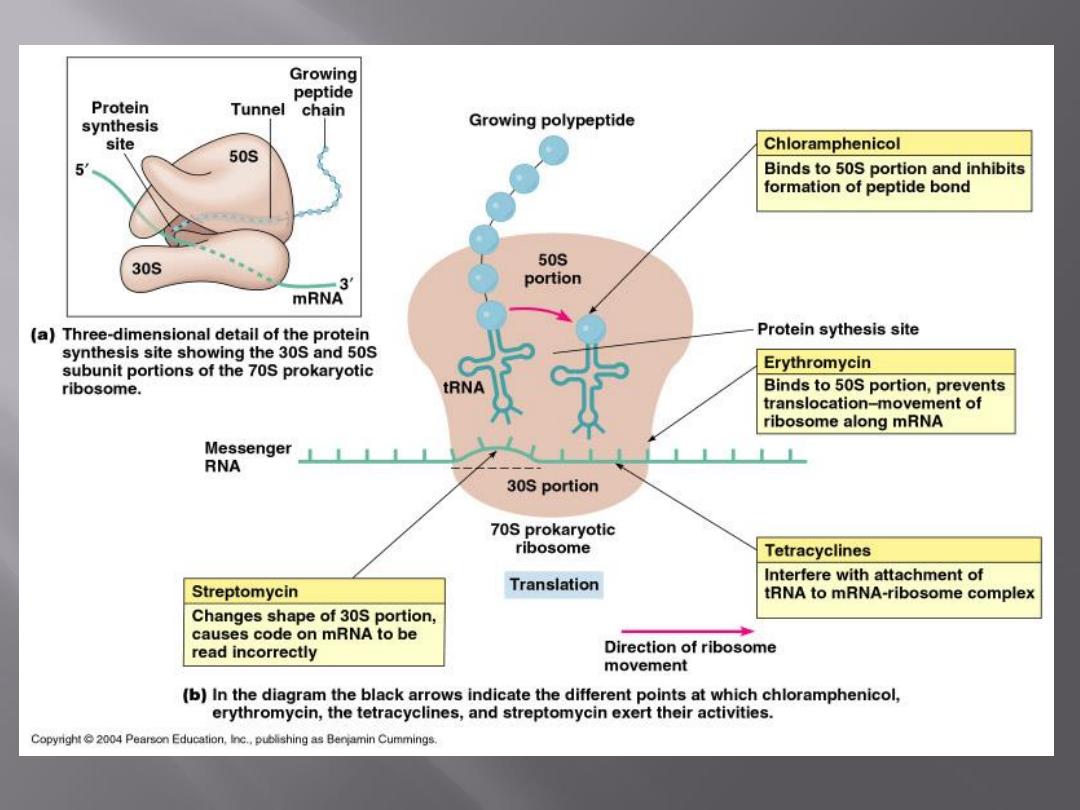
Tetracyclines bind to the 30S subunit of microbial
ribosomes. They inhibit protein synthesis by blocking
the attachment of charged aminoacyl-tRNA.
Thus, they prevent introduction of new amino acids to
the nascent peptide chain. The action is usually
inhibitory and reversible upon withdrawal of the drug.
Resistance
to
tetracyclines
occurs
by
three
mechanisms—efflux,
ribosomal
protection,
and
chemical modification. The first two are the most
important and occur as follows: Efflux pumps, located
in the bacterial cell cytoplasmic membrane, are
responsible for pumping the drug out of the cell.

Tet gene products are responsible for protecting the
ribosome, likely through mechanisms that induce
conformational changes. These conformational
changes either prevent binding of the tetracyclines
or cause their dissociation from the ribosome.
This is often plasmid-controlled. Mammalian cells
do not actively concentrate tetracyclines.

The glycylcyclines are synthetic analogues of the
tetracyclines.
The agent that is available for use in the United
States and Europe is tigecycline, a derivative of
minocycline.
The glycylcyclines inhibit protein synthesis in a
manner similar to the tetracyclines; however, they
are bactericidal, likely due to their more avid binding
to the ribosome.

Tigecycline is active against a broad range of gram-
positive and gram-negative bacteria, including
strains resistant to the typical tetracyclines.
The clinical activity of this agent is still undergoing
investigation, but currently its major use appears to
be in the treatment of skin and skin structure
infections and in intra-abdominal infections,
particularly caused by bacterial pathogens resistant
to a variety of other antimicrobial agents.

Chloramphenicol binds to the 50S subunit of the
ribosome. It interferes with the binding of new amino
acids to the nascent peptide chain, largely because
chloramphenicol inhibits peptidyl transferase.
Chloramphenicol is mainly bacteriostatic, and growth
of microorganisms resumes when the drug is
withdrawn.
Microorganisms resistant to chloramphenicol produce
the enzyme chloramphenicol acetyltransferase, which
destroys drug activity.
The production of this enzyme is usually under control
of a plasmid.

Quinupristin/dalfopristin is a combination
of two pristinamycin derivatives.
These two agents act synergistically to
achieve bactericidal activity against gram-
positive bacteria not seen with either agent
alone.
The mechanism of action appears to be
irreversible binding to different sites on the
50S ribosome.

The oxazolidinones are a relatively new
class of antimicrobial agents that possess a
unique mechanism of inhibition of protein
synthesis
primarily
in
gram-positive
bacteria.
These compounds interfere with translation
by inhibiting the formation of N-
formylmethionyl-tRNA,
the
initiation
complex at the 30S ribosome.
Linezolid is the agent that is currently
commercially available.

Examples of drugs acting by inhibition of nucleic
acid synthesis are the quinolones, pyrimethamine,
rifampin,
sulfonamides,
trimethoprim,
and
trimetrexate.
Rifampin inhibits bacterial growth by binding
strongly to the DNA-dependent RNA polymerase
of bacteria.
Thus, it inhibits bacterial RNA synthesis. Rifampin
resistance results from a change in RNA
polymerase due to a chromosomal mutation that
occurs with high frequency.

The mechanism of rifampin action on viruses
is different. It blocks a late stage in the
assembly of poxviruses.
All quinolones and fluoroquinolones inhibit
microbial DNA synthesis by blocking DNA
gyrase.
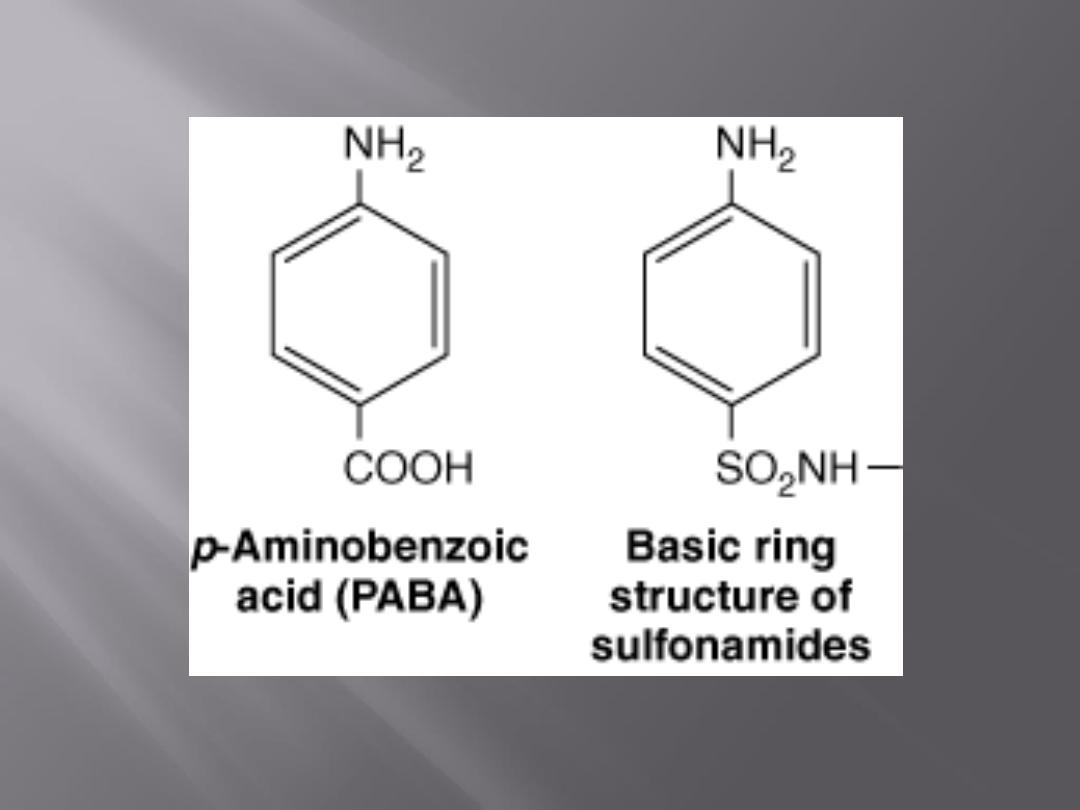
For many microorganisms, p-aminobenzoic
acid (PABA) is an essential metabolite.

The specific mode of action of PABA involves an
adenosine
triphosphate
(ATP)-dependent
condensation of a pteridine with PABA to yield
dihydropteroic acid, which is subsequently
converted to folic acid.
PABA is involved in the synthesis of folic acid, an
important precursor to the synthesis of nucleic
acids.
Sulfonamides are structural analogs of PABA and
inhibit dihydropteroate synthetase.

Sulfonamides can enter into the reaction in place of
PABA and compete for the active center of the
enzyme.
As a result, nonfunctional analogs of folic acid are
formed, preventing further growth of the bacterial
cell.
The inhibiting action of sulfonamides on bacterial
growth can be counteracted by an excess of PABA
in the environment (competitive inhibition).
Animal cells cannot synthesize folic acid and must
depend upon exogenous sources.

Some bacteria, like animal cells, are not inhibited by
sulfonamides.
Many other bacteria, however, synthesize folic acid
as mentioned above and consequently are
susceptible to action by sulfonamides.
Trimethoprim (3,4,5-trimethoxybenzylpyrimidine)
inhibits dihydrofolic acid reductase 50,000 times
more efficiently in bacteria than in mammalian
cells.

This
enzyme
reduces
dihydrofolic
to
tetrahydrofolic acid, a stage in the sequence leading
to the synthesis of purines and ultimately of DNA.
Sulfonamides and trimethoprim each can be used
alone to inhibit bacterial growth.
If used together, they produce sequential blocking,
resulting in a marked enhancement (synergism) of
activity.
If used together, they produce sequential blocking,
resulting in a marked enhancement (synergism) of
activity.
Such mixtures of sulfonamide (five parts) plus
trimethoprim (one part) have been used in the
treatment of pneumocystis pneumonia, malaria,
shigella enteritis, systemic salmonella infections,
urinary tract infections, and many others.

Pyrimethamine
also
inhibits
dihydrofolate
reductase, but it is more active against the enzyme
in mammalian cells and therefore is more toxic
than trimethoprim.
Pyrimethamine plus sulfonamide or clindamycin is
the current treatment of choice in toxoplasmosis
and some other protozoal infections.
