6
Lecture 3+4 - Heme-phenylalanine
metabolism
Hemoglobin metabolism
Heme synthesis
Step one:
The formation of α-aminolevulinate (ALA) from succinyl-
CoA (is derived from citric acid cycle) + glycine
This step occurs in the mitochondria.
Step two:
2 molecules of ALA are condensed to form Porpho-bilinogen
(PBG).
This step occurs in the cytosol.
Step three:
The formation of the tetrapyrrole: uro-porphyrinogen by
condensation of four molecules of PBG
Step four: The formation of protoporphyrin.
The last Step The Incorporation of Iron into Protoporphyrin
Porphyrias
Definition: are a group of genetic disorders of heme
metabolism due to abnormalities in the pathway of
biosynthesis of heme.
Cause: enzymic deficiency or blockage which could be
genetic or acquired.
Types:
Hepatic porphyrias The defect is primarily in the liver
Erythropoitic porphyrias
The defect is primarily in the bone marrow.
Signs and symptoms
1- Anemia
2- Recurrent abdominal pain.
3- Skin abnormalities and photosensitivity.
4- Inflammation of the nerve (neuritis).
5- Neuropsychiatric signs.
Biochemical findings:
1- Low Hb levels.
2- Increased Porphyrin products in the blood.
3- Excretion of porphyrines in urine as (Uroporphyrins) or in
the feces as (coproporphyrines) which may change color on
standing.
4- Enzymic studies.
Hemoglobinopathies
Definition: Are genetic diseases due to Hb molecule
abnormality in which an individual inherited the allele for an
abnormal hemoglobin from one or both parents.
The erythrocytes of these individuals are few and abnormal, In
addition to large numbers of immature cells.
Types: the most common are sickle cell anemia&thalassemias
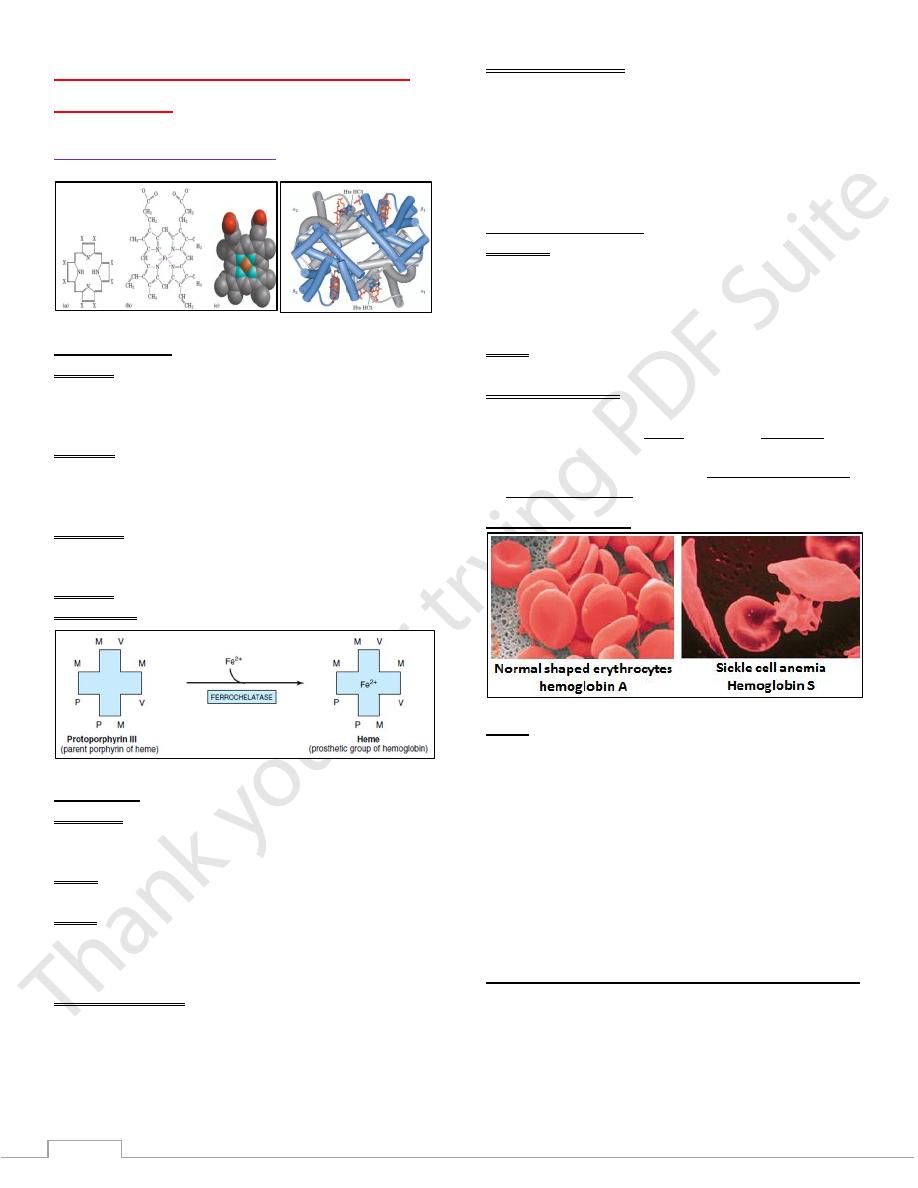
Sickle Cell Anemia
The altered properties of hemoglobin (S) result from a single
amino acid substitution, a Valine instead of a Glutamine at
position 6 in the two chains.
These cause deoxyhemoglobin S to be insoluble & forms
long, insoluble fibers characteristic of sickle shape of
RBCs
Abnormal hemoglobin
Types: there are two types:
The sickle-cell disease:
Individuals who receive the sickle-cell allele from booth
parents and are the homozygous for of the gene encoding
hemoglobin.
Sickle-cell trait:
Individuals who receive the sickle-cell allele from only one
parent. (heterozygous).
Thus heterozygous experience a milder condition , only about
1% of their erythrocytes become sickled on deoxygenation.
These individuals may live completely normal if they avoid
vigorous exercise or other stresses on the circulatory system.
Sickle-cell anemia is a life-threatening and painful disease.
People with sickle-cell anemia suffer from repeated crises
brought on by physical exertion, infection, respiratory dis.
They become weak, dizzy, and short breath, with an increased
pulse rate.
The hemoglobin content of their blood is only about half the
normal value.
7
Because sickled cells are very fragile and rupture easily; this
results in anemia.
A more serious consequence is that capillaries become
blocked by the long, abnormally shaped cells, causing severe
pain & interfering with normal organ function - a major factor
in the early death of many people with the disease.
Thalassemias
The genetic defects known as thalassemia result from: the
partial or total absence of one or more α or β chains of
hemoglobin.
Either the α chain (alpha thalassemia) or β chain (beta
thalassemia) can be affected.
There are two types of thalassemia:
Thalassemia major: in patients homozygous to the defect.
Is a more severe form of anemia, with splenomegaly and
abnormal bone marrow function.
Thalassemia minor: in patients heterozygous to the defect.
Is usually a symptom-free disease.
Treatment:
Apart from marrow transplantation, treatment is symptomatic.
Diagnosis of hemoglobinopathies
1) Family history and clinical examination.
2) Signs and symptoms:
Severe anemia, hemolytic crisis, splenomegaly, jaundice…
3) Laboratory findings:
Low Hb, raised serum bilirubin, increased bilirubin execration
in urine.
Abnormal blood and bone marrow films & Hb electrophoresis.
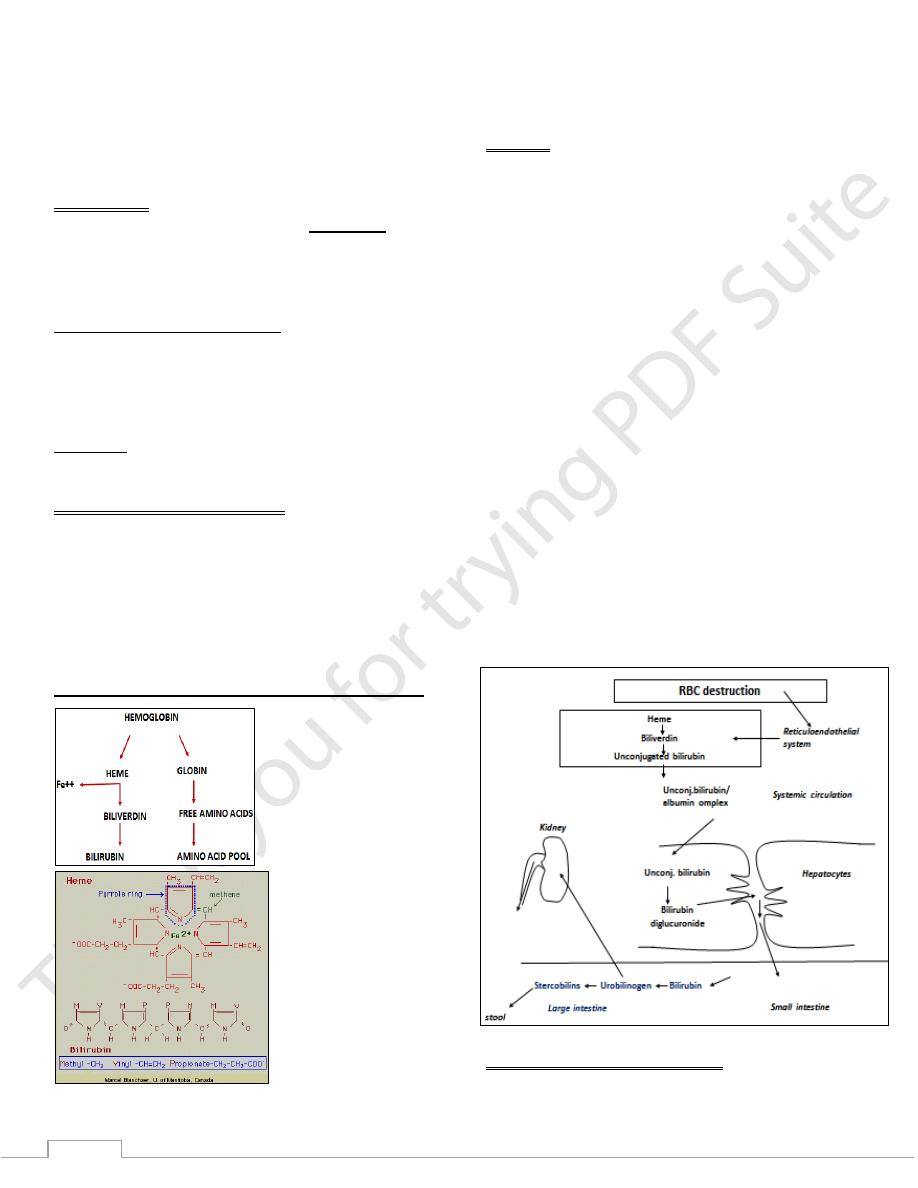
Hemoglobin catabolism and hyperbilirubinemia
biliverdin reductase reduces the methenyl bridge between
pyrrole III and pyrrole IV to a methylene group to produce
bilirubin, a yellow pigment.
Bilirubin
Bilirubin: is a yellowish chemical pigment results from the
catabolism of heme in the reticuloendothelial system.
Consists of four pyrrole rings linked by methane bridges in a
liner structure.
When first formed it is water insoluble and called: (indirect or
unconjugated) bilirubin.
Bilirubin formed in peripheral tissues is transported to the
liver by plasma albumin.
Metabolism of bilirubin occurs primarily in the liver. It can be
divided into three processes:
1) Uptake of bilirubin by liver parenchymal cells.
2) Conjugation of bilirubin with glucuronate in the
endoplasmic reticulum.
3) Secretion of conjugated bilirubin into the bile.
After conjugation with glucuronic acid, bilirubin becomes
water soluble and called: Direct or conjugated bilirubin.
It is estimated that 1 g of hemoglobin yields 35 mg of
bilirubin.
The daily bilirubin formation in human adults is
approximately 250–350 mg, deriving mainly from
hemoglobin but also from ineffective erythropoiesis and
from various other heme proteins such as cytochrome P450
The normal serum bilirubin con. Is below 1mg/100ml of
blood. If it is more than this, then hyperbilirubinemia
develops and could lead to jaundice.
Hyperbilirubinemia and jaundice
When bilirubin in the blood exceeds 1 mg/dL,
hyperbilirubinemia exists.

8
Causes
May be due to the production of more bilirubin than the
normal liver can excrete: This causes prehepatic jaundice.
Or it may result from the failure of a damaged liver to excrete
bilirubin produced in normal amounts: This causes hepatic
jaundice.
Obstruction of the excretory ducts will also cause
hyperbilirubinemia: this cause obstructive jaundice.
Jaundice
Is the yellowish discoloration of skin and mucous membranes
due to the deposition of bilirubin in the tissues.
The principle cause is the elevation of serum bilirubin above
the normal serum levels.
Neonatal “physiologic jaundice”
This transient condition is the most common cause of
unconjugated hyperbilirubinemia due to:
o Destruction of fetal Hb and replacement by adult Hb.
o It results from an accelerated hemolysis around the time of
birth and an immature hepatic system: for the uptake,
conjugation, and secretion of bilirubin
Appears during the 3
rd
to the 10
th
day of life.
Moderate anemia and hyperbilirubinemia.
The child looks normal, feeds well.
A slight skin and mucous membranes discoloration.
More common in premature children.
Urine and stool color is slightly deeper than normal.
Kernicterus, could develop in some sever cases which can
cause mental retardation.
Phenylalanine metabolism
Phenylalanine is an essential AA from which tyrosine is
formed by the action of the enzyme phenylalanine
hydroxylase.
Tyrosine is then used in the synthesis of many essential
metabolic products such as:
thyroid hormones, melanin (the main pigment for the skin
,eyes and hair), and adrenaline and dopamine
(dihydroxyphenylalanine) which are the main
neurotransmitters in the central nervous system.
Inherited enzymatic abnormalities in the pathway of the
AA phenylalanine will led to special clinical abnormalities:
Phenylketonuria:
This condition is caused by an abnormality of the
phenylalanine hydroxylase enz. System, which catalysis the
conversion of phenylalanine to tyrosine.
If this pathway is blocked or decreased then
Phenylalanine is converted into phenylketons and pyruvic acid
which then execrated in the urine together with phenylalanine.
On the other hand there will be a shortage or absence of
tyrosine in the body which will led to different pathologies.
The disease acquired its name from the recognition of this
phenyl Kenton in the urine.
The disease affects many neo-born babies causing brain
damage if not treated within few weeks.
Screening of neo-born infants is carried out in many countries
to detect cases early:
The urine is turned olive-green on the addition of FeCl3 to the
urine sample.
The clinical features are
1) Irritability
2) Feeding problems and vomiting.
3) Fits in the first few weeks of life.
Febrile fits: Encephalitis, meningitis, urinary tract infections
Respiratory tract infection.
4) Mental retardation develops between 4-6 months.
5) Generalized eczema in many cases.
6) Reduced melanin formation: many patients have a pale skin,
fair hair and blue eyes.
7) Babies exposed in utero to a high levels of phenylalanine of
diseased mothers may be mentally retarded and show other
congenital abnormality even though they are themselves are
not phenylketonuric (Maternal Phenylketonuria )
Management
The aim of management: is to lower blood phenylalanine
levels by giving a low phenylalanine diet.
Such treatment is difficult, expensive for the patient & parents.
Tyrosine must be included in the diet as it is the precursor of
many important metabolites.
Heterozygous Phenylketonuria is clinically normal but may be
detected by biochemical tests.

9
Alkaptonuria
Alkaptonuria: is an inherited deficiency of Homogentisic acid
oxidase results in excretion of large amounts of alkapton in
urine.
Homogentisic acid accumulates in blood, tissues.
Oxidation and polymerization of this substance produce the
pigment alkapton which has a deep black color similar to that
of melanin.
Deposition of these pigments in cartilages may cause joint
disease and may be visible as darkening of the ears.
Patients pass black urine if it is alkaline, or the urine will
be dark as it becomes more alkaline on standing.
These findings may be absent in significant number of
cases and often first noticed by the mother who is worried
about the napkins which only becomes black when
washed in alkaline soaps or detergents.
This condition is compatible with normal life span and
treatment is unnecessary but arthritis is common in old age
patients.
Heterozygote are not detectable by clinical or by biochemical
findings.
Late in the disease, there is artheritis and connective tissue
pigmentation due to oxidation of homogentisate to
benzoquinone acetate which polymerizes and binds to
connective tissue.
Albinism (Absence of melanin pigment)
A deficiency of tyrosinase in melanocytes causes albinism.
The patient lacked pigmentation in the skin hair and iris.
The eyes appear pink, acute photosensitivity occurs because of
lack of melanin pigment in the skin and eyes.