
Unit 1 - Immunology
44
Lecture 4+5+6 - Immunodeficiencies
Definition
It is a condition in which the immune system is failed to
protect the host from disease-causing agents or from
malignant cells.
Immunodeficiency disease results from the absence or
failure of normal function of one or more elements of the
immune system.
The immunodeficiencies should be suspected in every
patient, irrespective of age, who has recurrent, persistent,
sever or unusual infections.
Classification
A. Primary immunodeficiency: a condition results from a
genetic or developmental defect in immune system
(intrinsic defect). In such a condition, the defect is present
at birth although it may manifest itself later in life.
B. Secondary immunodeficiency or (acquired
immunodeficiency): is the loss of immune function and
results from exposure to various agents (disease or therapy).
The most common one is acquired immune-deficiency
syndrome or AIDS, which results from infection with the
human immunodeficiency virus 1 (HIV-1).
1) Primary immunodeficiency
Primary immunodeficiencies may affect either adaptive or
innate immune functions.
Specific immunodeficiency diseases involve
abnormalities of T or B cells, the cells of the adaptive
immune system.
Non-specific immunodeficiency diseases involve
abnormalities of elements such as complement proteins or
phagocytes, which act non-specifically in immunity..
Immunodeficiency diseases cause increased susceptibility
to infection in patients. The infections encountered in
immunodeficient patients fall into two categories:
Patients with defects in immunoglobulins, complement
proteins or phagocytes are very susceptible to recurrent
infections with encapsulated bacteria such as
Haemophilus influenzae, Streptococcus pneumoniae and
Staphylococcus aureus. These are called pyogenic
infections, because the bacteria give rise to pus formation.
On the other hand, patients with defects in cell-
mediated immunity, in T cells, are susceptible to
overwhelming, even lethal; infections with opportunistic
microorganisms include yeast and common viruses such
as chickenpox.
Defects in the lymphoid Lineage
A. Primary B-cell deficiencies include:
Patients with common defects in B-cell function have
recurrent pyogenic infections such as pneumonia, otitis
media and sinusitis
1) X- Linked Agammaglobulinemia (XLA) early
B- cell maturation fails
Affected males have few or no B cells in their blood or
lymphoid tissue; their lymph nodes are very small and
their tonsils are absent. Their serum usually contains no
IgA, IgM, IgD or IgE, and only small amounts of IgG
(less than 1 md/dl). Infants for the first 6 – 12 months of
life are protected from infection by the maternal IgG. As
this supply of IgG is exhausted, affected male develop
recurrent pyogenic infections.
The gene that is defective in X-LA is a B-cell cytoplasmic
tyrosine kinase (btk) belonging to the src oncogene family.
It encoded B- cell signal transduction molecule called
Burton’s tyrosin kinase is obviously vital for the process of
B-cell maturation. Bone marrow of males with X-LA
contains normal numbers of pre-B cells but, as a result of
mutations in the btk gene, they cannot mature to B cells.
Treatment by periodic Intravenous administration of Igs
Unit 1 - Immunology
45
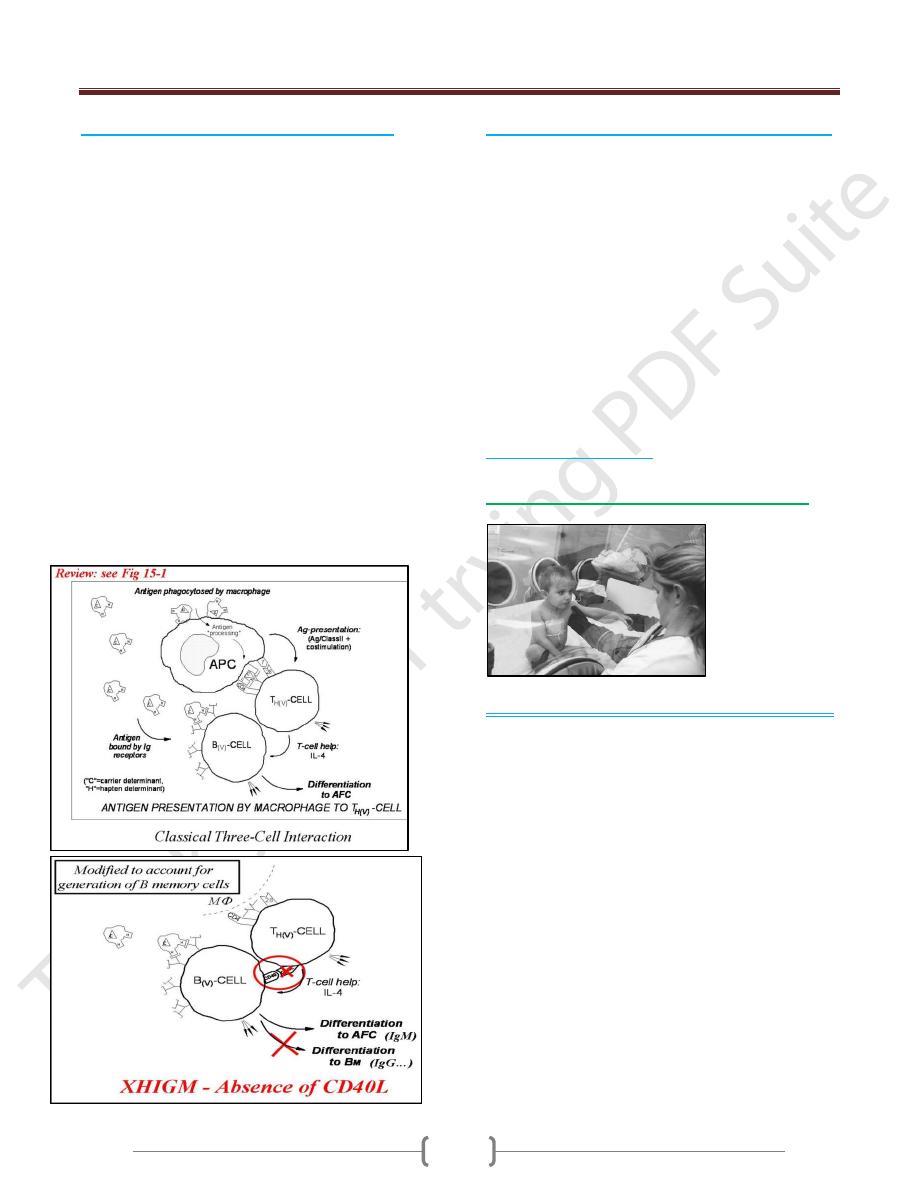
2) X- Linked Hyper IgM Syndrome (XHM)
In XHIgM the B cells cannot make the switch from IgM
to IgG, IgA and IgE synthesis that normally occurs in B-
cell maturation.
As a result, patients have decreased levels of serum IgG
and IgA and elevated levels of IgM some times as high as
10 mg/ml (normal Igm concentration is 1.5 mg/ ml).
It results from a variety of genetic defects that affect the
interaction between T-lymphocytes and B-lymphocytes.
It is inherited as an X- linked recessive disorder.
In normal B cells, this switch to IgE is induced by two
factors:
IL-4 must bind to the B-cell receptor for IL-4, and
The CD40 molecule on the B-cell surface must bind to
the CD40 ligand on activated T cells.
70% is due to defect in the gene encoding the CD40
ligand (CD40L) on the membrane of TH cells
Children in first two years suffer recurrent infections,
especially respiratory infections caused by opportunistic
pathogens.
Treatment by administration of intravenous Ig.
3) Common variable immunodeficiency (CVID)
There are defect in T cell signaling to B cells.
Individuals with CVID have acquired
agammaglobulinaemia in the second or third decade of
life, or later. Both males and females are equally affected
and the cause is generally not known, but may follow
infection with viruses such as Epstein – Barr virus (EBV).
Patients with CVID, like males with X-LA, are very
susceptible to pyogenic organisms.
Most patients (80%) with CVID have B cells that do not
function properly and are immature. The B cells are not
defective; instead, they fail to receive proper signals from
the T cells.
Patients with CVID should be treated with intravenous
gammaglobulin.
4) Selective IgA deficiency
B. Primary T-cell deficiencies include:
1) Severe Combined Immunodeficiency (SCID):
Infants with SCID have very few lymphocytes in their
blood (fewer than 3000/ml). Their lymphoid tissue also
contains few or no lymphocytes. The thymus has a fetal
appearance.
Patients with no T cells, or poor T-cell function, are
susceptible to opportunistic infections. Since B-cell
function in humans is largely T-cell dependent, T-cell
deficiency also results in humoral immunodeficiency.
The infants have prolonged diarrhea due to rotavirus or
bacterial infection of the GIT, and develop pneumonia
usually due to protozoal infection.
The common yeast organism Candida albicans grows in
the mouth or on the skin of the patients with SCID.
If the patients with SCID are vaccinated with live
organisms such as poliovirus or BCG they die from
progressive infection with these organisms

Unit 1 - Immunology
46
1. Over 50% of cases are caused by a gene defect on the X
chromosome. SCID is more common in males than
females infants (3:1)
Genetic defect in γ-chain of the IL-2R also shard
receptors for other cytokines IL-4, 7, 11, and 15.
2. The remaining cases of SCID are due to recessive genes
on other chromosomes of these, half have a genetic
deficiency of adenosine deaminase (ADA) or purine
nucleoside phosphorylase (PNP), resulting in the
accumulation of metabolites that are toxic to lymphoid
stem cells. These metabolites inhibit the enzyme
ribonucleotide reductase, which is required for DNA
synthesis and for all replication.
3. Other autosomal recessive form of SCID results from a
mutation in either of the recombinase- activating genes
encoding RAG-1 or RAG-2.
These two genes are absolutely required for cleaving
double- stranded DNA before recombination of DNA
to form the immunoglobulin genes and the genes
encoding the lymphocyte cell receptor that
characterized mature B and T cells.
If these gene rearrangements do not occur, B and T
cells do not develop.
The optimal treatment: is a bone marrow transplant from a
completely histocompatible donor, usually normal sibling,
or the affected infants die within the first 2 years of life.
Also gene therapy of RAG
2) The DiGeorge Syndrome (Congenital Thymic
Aplasia)
The thymic epithelium is derived from the third and
fourth pharyngeal by the sixth week of human gestation.
Defect is associated with the deletion in the embryo of a
region on chromosome 22
The T-cell deficiency is variable, depending on how badly
the thymus is affected. Affected infants have distinctive
facial features in that their eyes are widely separated.
They also have congenital malformations of the heart or
aortic arch and neonatal tetany from the hypoplasia or
aplasia of the parathyroid glands.
Treatment is by supportive therapy, or thymic
epithelial transplant.
3) Wiskott- Aldrich Syndrome (WAS):
WAS is an X-linked immunodeficiency disease.
Affected males have small and abnormal platelets, which
are also few in numbers (thrombocytopenia) which may
lead to fatal bleeding. Boys with WAS develop severe
eczema as well as pyogenic and opportunistic infections.
Their serum contains increased amounts of IgA & IgE,
normal levels of IgG and decreased amounts of IgM.
Their T cells are defective in function. This fails to
occur in the WAS, with the result that collaboration
among immune cell is faulty.

Unit 1 - Immunology
47
C. Genetic Defect in Complement Proteins
Deficiencies of the classical pathway components, C1q,
C1r, C1s, C4, or C2 results in susceptibility to develop
immune complex disease such as systemic lupus
erythematosus (SLE). This correlates with the known
function of the classical pathway in the dissolution of
immune complexes.
Deficiencies of C3, and the alternative pathway
components, factor H, or factor I result in increased
susceptibility to pyogenic infection; this correlates with
the important role of C3 in opsonization of pyogenic
bacteria.
Deficiencies of the terminal components C5-8, and of
the alternative pathway components, factor D and
properdin results in remarkable susceptibility to
infection with two pathogenic spp. Of Neisseria,
gonorrhoeae, and meningitides.
All these genetic complement component deficiencies
are inherited.
Treatment usually maintained with antibiotics.
Hereditary angioneurotic edema (HAE) is
due to C1 inhibitor deficiency
It is well-known disease resulted due to deficiency of the
complement system C1 inhibitor. This molecule is
responsible for dissociation of activated C1, by binding to
C1r2 and C1s2.
This disease is inherited as an autosomal dominant
trait
C1 inhibitor deficiency may be acquired later in life. In
some cases an autoantibody to C1inhibitor is found.
Patients with HAE have recurrent episodes of swelling of
various parts of the body (angioedema). When the edema
involves the intestine, abdominal pains ad cramps results,
with severe vomiting.
When the edema involves the upper airway, the patients
may choke to death from respiratory obstruction.
Angioedema of the upper airway therefore presents a
medical emergency, which requires rapid action to restore
normal breathing.
D. Defects in phagocytes
Phagocytic cells – polymorphonuclear leucocytes and
cells of the monocyte /macrophage lineage – are
important in host defense against pyogenic bacteria and
other intracellular microorganisms. A severe deficiency
of polymorphonuclear leucocytes (neutropenia) can result
in overwhelming bacterial infection.
Two genetic defects of phagocytes are clinically
important in that they result in susceptibility to severe
infections and are often fatal includes:
1. Chronic granulomatous disease
2. Leucocyte adhesion deficiency.
1) Chronic granulomatous disease (CGD) is due
to a defect in the oxygen reduction pathway
Is a genetic disease, 70% is an X- linked form
Defect in the ability of macrophages and PMNs to kill
phagocytosed organisms
Decrease in the ability of macrophages to serve as APCs.
Patients with CGD have defective NADPH oxidase
which catalyzes the reduction of O
2
to •O
2
by the
reaction:
NADPH + 2O
2
→ NADP
+
+ 2•O
2-
+ H
+
Thus, they are incapable of forming superoxide anions
(•O
2
) and hydrogen peroxide in their phagocytes,
following ingestion of microorganisms and so cannot
readily kill ingested bacteria or fungi organisms.
As a result, microorganisms remain alive in phagocytes of
patients with CGD. This gives rise to a cell-mediated
response to persistent intracellular microbial antigens, and
granulomas form. Children with CGD develop
pneumonia, infections the lymph nodes (lymphadenitis),
and abscesses in the skin, liver and other viscera.
Treatment with antibiotics
2) Leukocyte Adhesion Deficiency (LAD)
The receptor in the phagocyte membrane that binds to
C3b on opsonized microorganisms is critical for the
ingestion of bacteria by phagocytes. This receptor, an
integrin called complement receptor 3 (CR3), is
deficient in patients with LAD and consequently they

Unit 1 - Immunology
48
develop severe bacterial infections, particularly of the
mouth and GIT.
CR3 is composed of two polypeptide chains: an α chain
and β chain. In LAD, there is a genetic defect of the β
chain of CR3, encoded by a gene on chromosome 21.
Other integrin proteins share the same β chain, namely
lymphocyte function associated antigen (LFA-1) .
Genetic defect of LFA-1 leading to impairment of
adhesion of leukocytes to vascular endothelium and limits
recruitment of cells to sites of inflammation.
LAD varies in its severity; some affected individuals die
within a few years, whereas others may survive into their
forties.
2) Secondary or Acquired
Immunodeficiency
It results from exposure to a number of chemical &
biological
agents that induce immunodeficient state.
A. Drugs:
Corticosteroids: commonly used for treatment of
autoimmune disorders interfere with the immune response
in order to relief the disease symptoms
Immunosuppressive drugs, such as cyclosporine- A
used in transplantation patients which block the immune
attack to transplanted organ.
Cytotoxic drugs or radiation treatments given to treat
various forms of cancer frequently damage the dividing
cells in the body (depression hematopoiesis)
B. Nutrient Deficiency
Lymphoid tissues are very vulnerable to the damaging
effects of malnutrition.
Numerous enzymes with key roles in immune processes
required zinc, iron, vitamin B6, and other
micronutrients including selenium, and copper.
Lymphoid atrophy is a prominent morphological feature
of malnutrition (T selective deficiencies)
C. Infection:
Acquired Immune Deficiency Syndrome (AIDS):
The most significant global cause of immunodeficiency is
HIV infection. Over 25 million people have died from
AIDS since the first cases were described in 1981. As the
end of 2004, WHO estimate that, approximately 40
million people are living with HIV infection worldwide,
with approximately 5 million new infections and 3 million
deaths each year.
Acquired Immune Deficiency Syndrome (AIDS)
AIDS: Is caused by Human Immunodeficiency Virus
(HIV) a retrovirus, which is found in all cases of the
disease.
The primary targets of HIV are activated CD4+ T helper
lymphocytes but the virus can also infect several other
cell types including macrophages.
Infection leads to loss of T4 helper lymphocytes and
immunosuppression in the patient and the consequent
fatal opportunistic infections.
HIV is a lentivirus, a class of retrovirus.
The name lentivirus means slow virus, so called because
these viruses take a long time to cause overt disease.
Most lentiviruses target cells of the immune system and
thus disease is often manifested as immunodeficiency
Unit 1 - Immunology
49
There are two types of HIV: HIV-1 and HIV-2. These
cause clinically indistinguishable disease, although the
time to disease onset is longer for HIV-2.
The worldwide epidemic of HIV and AIDS is caused
by HIV-1 while HIV-2 is mostly restricted to west Africa
CD4 antigen is the main receptor for the virus entry, and
is present on CD4+ T lymphocytes and monocytes.
The binding of the viral envelope glycoprotein gp120 to
CD4 antigen results in conformational changes in gp120
that expose binding sites for chemokine receptors, which
serve as co-receptors for viral entry, these includes CCR5
and CXCR4
The disease is appeared at the first time in 1981, as clusters
of cases of Kaposi's sarcoma were reported in young
patients in San Francisco and New York. This was an
unusual occurrence since, in the United States, Kaposi's
sarcoma was a rare disease that normally occurred in
elderly men of Jewish or Mediterranean ancestry. however,
these new clusters of patients were all young male
homosexuals and the disease was much more aggressive
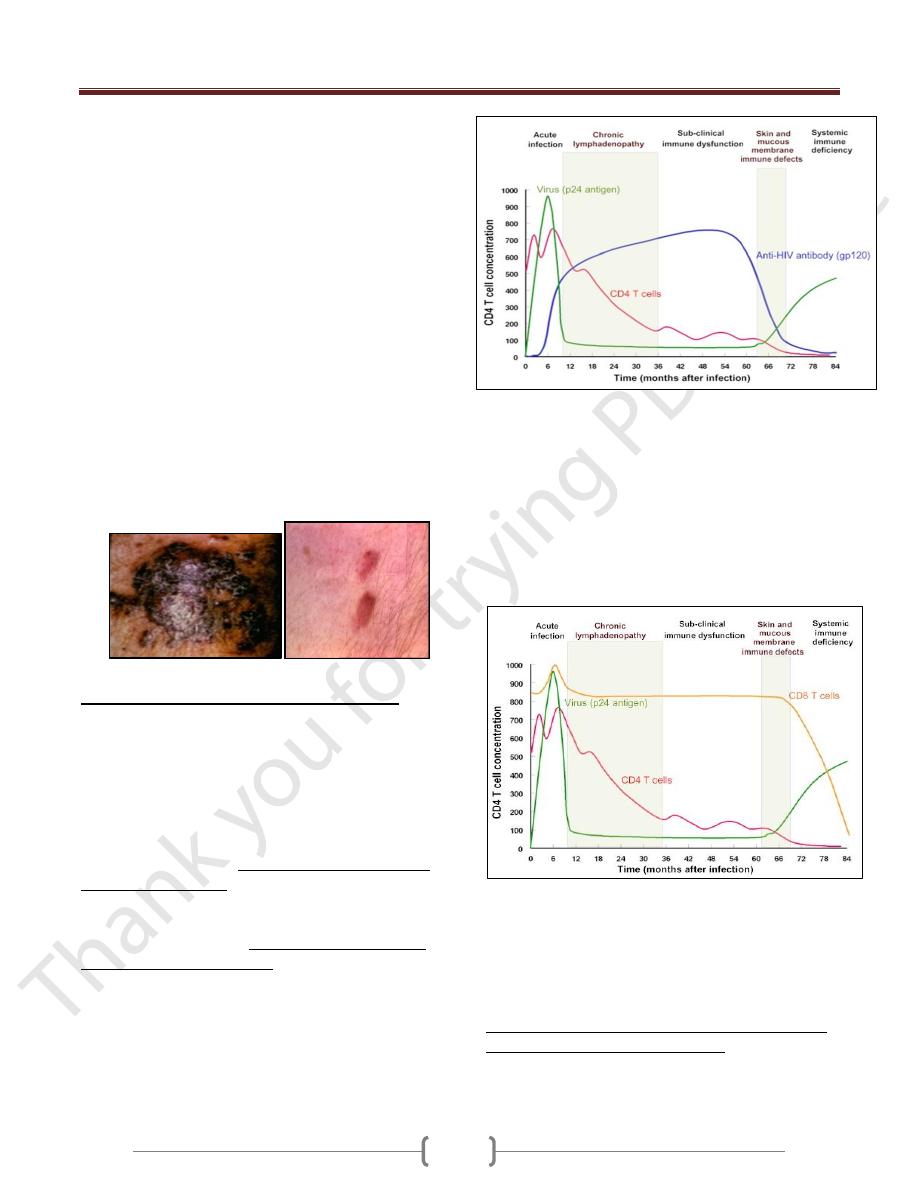
Immune Responses against HIV infection
Cell-mediated and humoral anti-HIV immune defense:
Cytotoxic T and B lymphocytes mount a strong defense
and virus largely disappears from the circulation.
Virus titer, CD4T cells and anti-gp120 titer during the
HIV infection
After the increased cell-mediated immune response, there
is a rise in antibodies in the serum of infected individuals
2-3 weeks after infection, but though these lack the ability
to inhibit viral infection. During this period, more than 10
billion new HIV particles are produced each day. They are
rapidly cleared by the immune system especially anti-
HIV antibody (gp120). So neutralizing antibodies play a
role in controlling HIV viremia.
Despite the presence of high numbers of HIV specific
CTLs in the peripheral blood, but like antibody
responses unable to eliminate infection.
At this stage, most of this virus is coming from
recently infected proliferating CD4
+
cells. Thus, the
virus is destroying the cells that are proliferating.
The infected cells that are producing this virus are
destroyed either by the immune system or by the virus
and have a half-life about 1 day.
Although activated, proliferating CD4+ cells are
destroyed by the immune system, a small fraction of the
infected cells survive long enough to revert back to the
resting memory state (as do non-infected CD4
+
memory cells).
The resting memory cells do not express viral antigens but
do carry a copy of the HIV genome which remains latent
until the cells are reactivated by antigen. These memory
cells may survive many years and constitute a
reservoir that is very important in drug-based
therapy.
Unit 1 - Immunology
50
During this period, the virus disseminates to other regions
including to lymphoid and nervous tissue. This is the most
infectious phase of the disease.
Loss of CD4
+
cells & collapse of the immune
response
During the course of infection, there is a profound loss of
the specific immune response to HIV because:
Responding CD4+ cells become infected. Thus, there is
clonal deletion leading to tolerance and escape of HIV
from the immune surveillance.
Activated CD4+ T cells are susceptible to apoptosis.
Spontaneous apoptosis of uninfected CD4
+
and CD8
+
T
cells occurs in HIV-infected patients.
Also there appears to be selective apoptosis of HIV-
specific CD8
+
cells
the number of follicular dendritic cells falls over time,
resulting in diminished capacity to stimulate CD4+ cells
More severe infections are associated with a low
CD+4 T cell count
It is the phase of the disease that lacks the neoplasms and
opportunistic infections that are the definition of AIDS
Patients at this stage of the disease show weight loss and
fatigue together with fungal infections of the mouth,
finger and toe nails especially with Candida
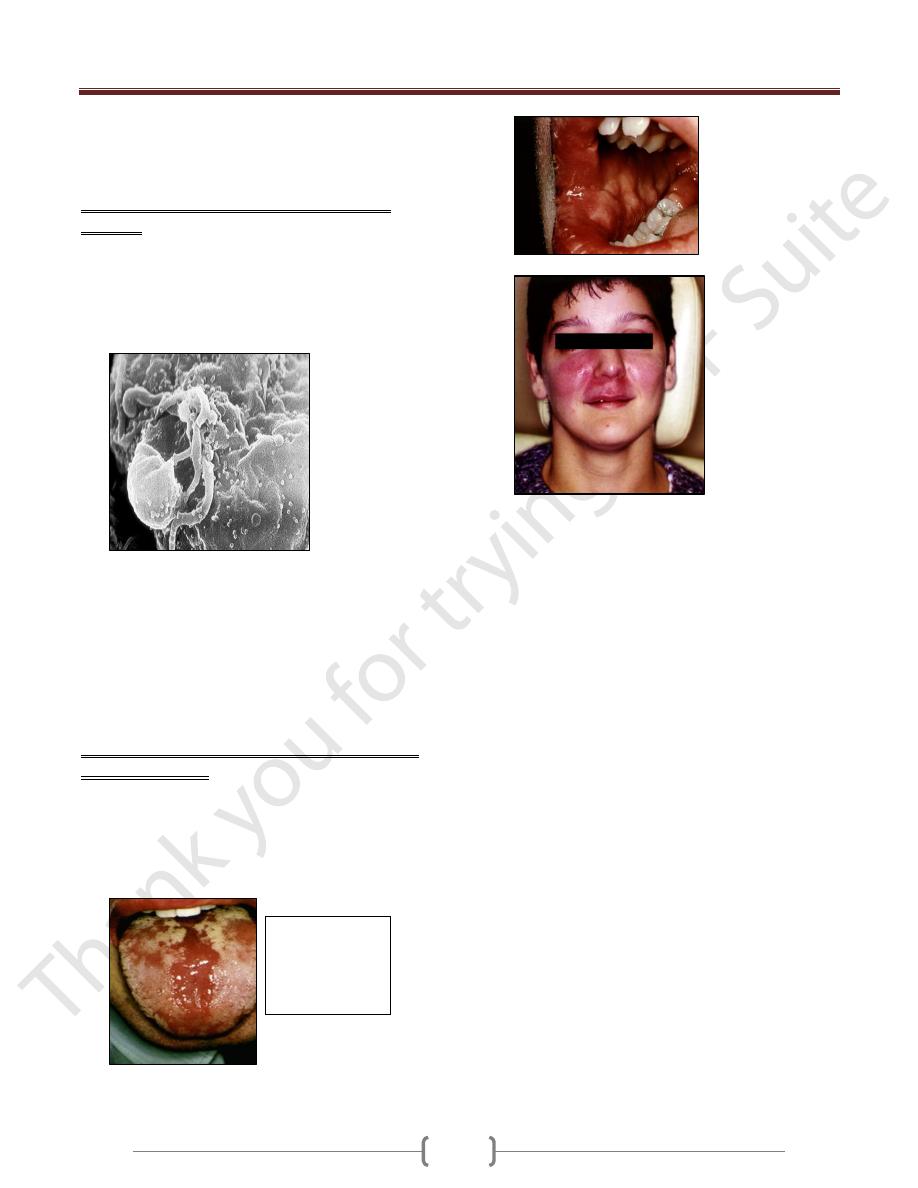
Orofacial
granulomatos
is with cobble
stone mucosa
in AIDS
Facial
sarcoidosis
in AIDS
Opportunistic
infections that
are the definition
of AIDS