Unit 11: Hematopoietic and Lymphoid Systems
156
Haemopoiesis
Haemopoiesis is process of blood cell formation, starts
with a stem cell that can give rise to the separate cell
lineages. Haemopoiesis include:
Erythropoiesis (formation of red cells).
Myelopoiesis (formation of granulocytes and monocytes).
Thrombopoiesis (formation of platelets).
Haemoglobin (Hb) is iron containing metalloprotein
found in the red blood cells. The main function of
haemoglobin is to carry O
2
to the tissues and to return
carbon dioxide (CO
2
) from the tissues to the lungs, Hb
molecule consist of:
4 polypeptide chains.
4 haem group (each composed of Fe
2+
& portoporphyrin)
RED CELL DISORDERS
Anemia in general
Anemia defined as a reduction in the haemoglobin
concentration of the blood less than 13.5 g/dL in adult
males, less than 11.5 g/dL in adult females.
Anemia
caused by:
Blood loss (hemorrhage).
Increased red cell destruction (hemolysis).
Decreased red cell production.
Symptoms
Shortness of Breath.
Weakness.
Palpitation.
Headaches.
Heart failure (especially in elderly).
Angina pectoris (especially in elderly).
Visual disturbances (very severe anemia).
Signs
General Signs
Are associated with all type of anemia include:
o Pallor of mucous membranes.
o Tachycardia.
o Cardiomegaly and a systolic flow murmur (especially
in elderly).
o Retinal haemorrhages (in severe anemia)
Specific Signs
Are associated with particular types of anemia e.g.:
o Koilonychia (spoon nails) with iron deficiency.
o Jaundice with hemolytic or megaloblastic anemia.
o Leg ulcers with sickle cell and hemolytic anemia.
o Bone deformities with thalassaemia major.
Classification
The most useful classification is that based on red cell
volume (MCV) and divides the anemia into:
Microcytic (MCV <80fL).
Normocytic (MCV 80 - 95fL).
Macrocytic (MCV >95fL).
Hemolytic anemias
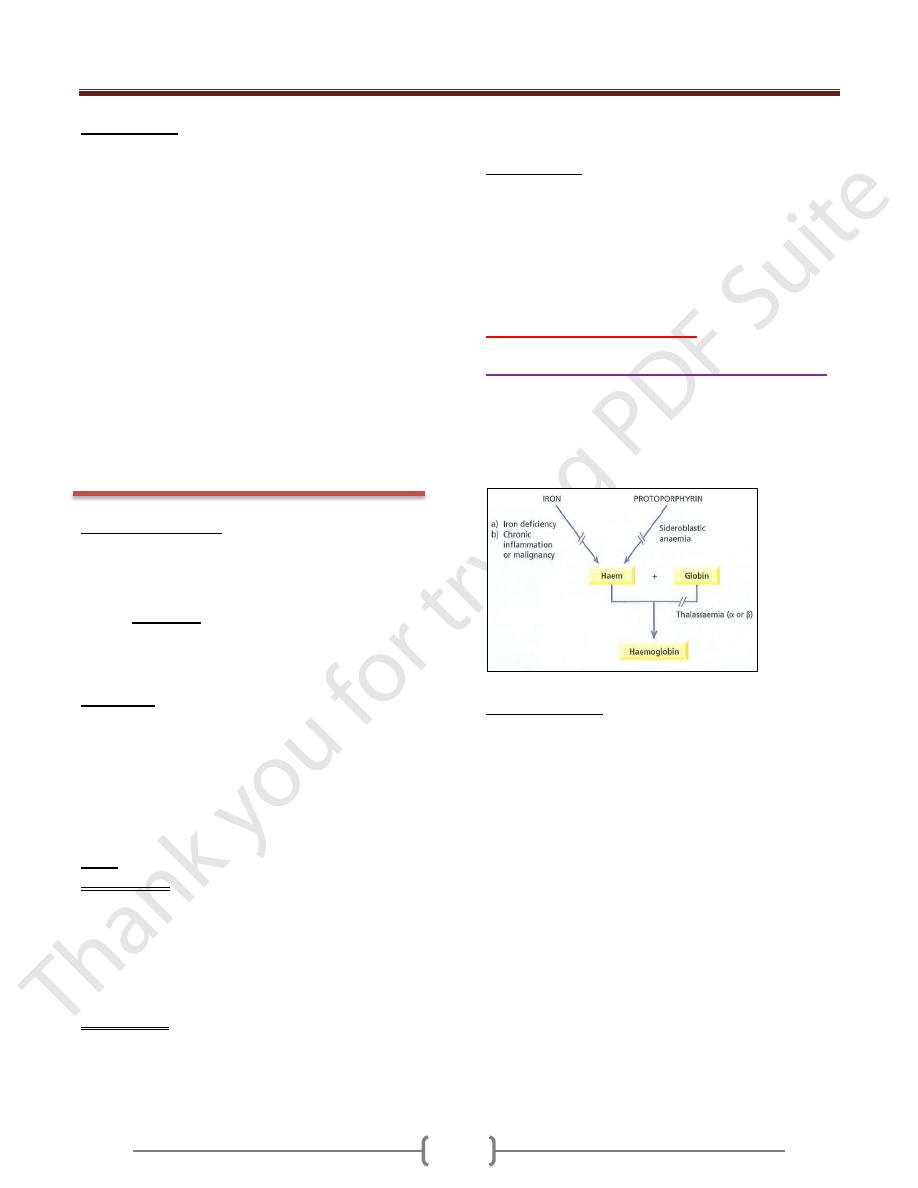
Microcytic anemia differential diagnosis:
1. Iron deficiency anemia.
2. Anemia of chronic disease (some cases).
3. Thalassaemia (major and minor)
4. Sideroblastic anemia (some cases)
Iron metabolism
Approximately 1 mg of iron is absorbed (in duodenum
and upper jejunum) and 1 mg lost (sweating, epidermal
shedding) each day.
Transferrin is the main iron carrier in plasma, transfer iron
from GIT cells to erythroblast (Hb forming cells) or store.
Iron stored in reticuloendothelial system (bone marrow,
liver and spleen) in 2 forms: ferritin and hemosiderin.
Serum iron measure iron bound to transferrin while total
iron binding capacity (T.I.B.C) measure transferrin not
bound to iron i.e. free transferrin.
Serum transferrin receptor (sTfR) is portion of the
transferrin receptor (found on surface of cells) that shed
into serum. The sTfR is regarded as a more stable marker
of iron store than ferritin in inflammatory state (since
ferritin is an acute phase reactant protein that increased in
inflammation).
Total amount of body iron is 2-6 g distributed in form of
Hb, myoglobin, ferritin, hemosiderin, transferrin bound
iron and enzymes cytochrome).

Unit 11: Hematopoietic and Lymphoid Systems
157
Iron deficiency Anemia
It is the most common cause of anemia worldwide. The
reticuloendothelial iron stores become completely
depleted before anemia occurs.
Causes of iron deficiency:
Chronic blood loss especially uterine or from the
gastrointestinal tract (most common cause).
Increased demands during infancy, adolescence,
pregnancy, lactation and in menstruating women.
Malabsorption or poor diet (rare) e.g. Gluten-
induced enteropathy.
Clinical features
General symptoms and signs of anemia
Specific symptoms and signs as:
1) Painless glossitis.
2) Angular stomatitis.
3) Koilonychia (spoon nails).
4) Dysphagia as a result of pharyngeal webs (Paterson
Kelly or Plummer-Vinson syndrome).
5) Pica (unusual dietary cravings e.g. earth, clay, ice).
6) Irritability, poor cognitive function and a decline in
psychomotor development (in children).
Laboratory findings:
Low Hb (Mild, moderate or severe anemia).
MCV < 80fL (Microcytic) & MCH (Hypochromic) <
27pg/L.
Blood film shows hypochromic microcytic cells with
target cells and pencil shaped poikilocytes.
Iron Studies used to confirm iron deficiency anemia,
include: Serum iron decreased; Total iron binding
capacity (TIBC) increased; Serum transferrin receptor
increased and Serum ferritin is very low.
Bone marrow iron (reticuloendothelial store) is absent
Treatment
Treat the underlying cause.
Iron replacement therapy (oral iron or parentral) 4 – 6
months to treat anemia and replenish iron store.
Anemia of chronic disorders
Common anemia occurs in patients with chronic
inflammatory and malignant diseases. The pathogenesis
of this anemia related to decreased release of iron from
macrophages (store) to plasma.
Laboratory findings:
Low Hb (mild and non-progressive anemia).
MCV and MCH Normal or slightly reduced (MCV
rarely <75 fL).
Blood film shows normochromic, normocytic or
mildly hypochromic microcytic.
Iron Studies include: serum iron decreased; TIBC
decreased; serum transferrin receptor normal and
serum ferritin is normal or increased.
Bone marrow storage (reticuloendothelial) iron is normal.
Treatment
Treat The underlying cause (does not respond to iron therapy).
Thalassaemia Trait
Group of genetic disorders that result in a reduced rate of
synthesis of globin chains (reduce Hb synthesis).
Laboratory findings:
Hb normal or low (absent or mild anemia).
MCV and MCH reduced (MCV ≤ 70 fL).
Red cell count is usually over 5.5 x 10
12
/L.
Blood film shows hypochromic microcytic red cells.
Iron Studies in thalassaemia minor are Normal.
Hb electrophoresis raised Hb A2.
Sideroblastic Anemia
It is a refractory anemia caused by defect in haem
synthesis (failure to use iron in Hb synthesis despite the
availability of high iron stores). Sideroblastic anemia
either congenital or acquired e.g. Lead poisoning.
Laboratory findings:
Blood film shows hypochromic microcytic red cells.
Iron Studies: Serum iron increased; TIBC normal; Serum
transferrin receptor normal; Serum ferritin increased.
Bone marrow iron is increased.
Presence of many pathological ring sideroblasts in
the bone marrow (diagnostic for sideroblastic anemia)
Macrocytic Anemia
In macrocytic anemia, the red cells are abnormally large
(mean cell volume MCV > 95 fL). Causes of
macrocytosis can be subdivided into 2 groups:
Megaloblastic: erythroblasts in the bone marrow show
delayed maturation of the nucleus relative to the cytoplasm
(asynchronous maturation) due defect in DNA synthesis.
Nonmegaloblastic erythroblasts in the bonemarrow are normal
Megaloblastic Anemia
Causes of megaloblastic anemia:
1) Vitamin B
l2
deficiency.
2) Folate deficiency.

Unit 11: Hematopoietic and Lymphoid Systems
158
3) Abnormalities of vitamin B
12
or folate metabolism
(nitrous oxide, antifolate drugs).
4) Defects of DNA synthesis (e.g. orotic aciduria, alcohol).
Causes of non megaloblastic macrocytosis:
1) Alcohol.
2) Liver disease (especially alcoholic).
3) Reticulocytosis (hemolysis or hemorrhage).
4) Aplastic anemia or red cell aplasia.
5) Hypothyroidism.
6) Myelodysplasia.
7) Myeloma and macroglobulinaemia.
8) Leucoerythroblastic anemia.
9) Myeloproliferative disease.
10) Pregnancy.
11) Newborn.
12) Congenital dyserythropoietic anemia (type II).
Vitamin B
12
(Cobalamin)
The vitamin B
12
is found in animal product only such as
liver, meat and dairy produce. Minimal adult daily
requirement is 1-2/µg. Body stores about 2-3 mg
(sufficient for 2-4 years).
Bl2 (in normal diet) is combined with the glycoprotein
intrinsic factor (IF) (which is synthesized by the gastric
parietal cells) in stomach. The IF-B
12
complex can then
bind to IF receptor in the distal ileum where B
l2
is
absorbed and IF destroyed.
Transcobalamin (TC) is vitamin B
l2
carrier in blood which
delivers B
l2
to bone marrow and other tissues.
Vitamin B
12
is a coenzyme for two biochemical reactions
in the body:
1) Methylation of homocysteine to methionine.
2) Conversion of methylmalonyl CoA to succinyl CoA.
Folate
Folate is found in liver, greens and yeast. Minimal adult
daily requirement is 100-150/µg. Body stores 10-12 mg
(sufficient for 4 months).
Dietary folates absorbed through duodenum and jejunum
(after conversion to methyltetrahydrofolate THF).
Folates are needed in a variety of biochemical reactions in
the body such:
1) Amino acid interconversions.
2) Synthesis of DNA purine precursors.
Both B
12
& folate deficiency cause defect in DNA synthesis
Causes of vitamin B
12
(Cobalamin) deficiency:
1) Nutritional (especially vegans)
2) Malabsorption:
Gastric causes (e.g., pernicious anemia, congenital lack or
abnormality of intrinsic factor, total or partial gastrectomy).
Intestinal causes (e.g. intestinal stagnant loop syndrome,
chronic tropical sprue, ileal resection and crohn's disease,
fish tapeworm).
Causes of folate deficiency:
1) Nutritional (especially old age, poverty, famine)
2) Malabsorption (tropical sprue, gluten induced enteropathy,
extensive jejunal resection or crohn's disease).
3) Excess utilization (pregnancy, lactation, hemolytic anaemias,
myelofibrosis, malignant disease, inflammatory diseases).
4) Excess urinary folate loss (active liver disease, congestive
heart failure)
5) Drugs (anticonvulsants, sulfasalazine).
6) Alcoholism.
Clinical features of megaloblastic anemia:
1) Symptoms of gradual, progressive anemia but sometimes
patients are asymptomatic (detected through the finding of
a raised MCV on a routine blood count).
2) Symptoms associated with vitamin B
l2
or Folic acid deficiency:
Peripheral neuropathy (sub-acute combined degeneration
of the cord) in severe B
12
deficiency only.
Gastrointestinal symptoms (due to vitamin B
l2
and folic
acid deficiency):
Anorexia and weight loss.
Diarrhea or constipation.
Glossitis (red, sore, smooth tongue)&angular cheilosis
Jaundice (due to destruction of red cell precursors in the
bone marrow).
Bruising (due to thrombocytopenia) and reversible
melanin skin hyperpigmentation.
Infections due to low WBC count (e.g. respiratory or
urinary tracts).
General tissue effects of B
12
& folate deficiencies:
1) Epithelial surfaces cells show macrocytosis, with
increased numbers of multinucleate and dying cells.
2) Infertility is common in (both men and women) and
recurrent fetal loss.
3) Folate or B12 deficiency in the mother predisposes to
neural tube defect in the fetus.
4) Cardiovascular disease (e.g. ischemic heart disease,
cerebrovascular disease or pulmonary embolus).
5) Prophylactic folic acid in pregnancy has been found to
reduce the subsequent incidence of acute lymphoblastic
leukemia in childhood.

Unit 11: Hematopoietic and Lymphoid Systems
159
Laboratory findings
A. Diagnosis megaloblastic anemia
1) Complete blood pictures:
Low Hb.
MCV > 95 fL (macrocytic).
Low WBC count (leucopenia)
Low platelet counts (thrombocytopenia).
Low reticulocyte count.
2) Blood film:
Macrocytic red cells (typically oval in shape).
Hyper segmented neutrophils nuclei (with 6 or more
lobes).
3) Bone marrow aspirate:
Hypercellular bone marrow.
Increased erythroid /myeloid ratio.
Erythroid cell precursors are large (megaloblastic) and
show failure of nuclear maturation but normal cytoplas
hemoglobinization (asynchronous maturation).
Myeloid cell changes (giant metamyelocytes).
Megakariocytes are decreased and show abnormal
morphology
4) Biochemistry:
elevation of serum unconjugated bilirubin.
elevation of lactate dehrogenase (LDH).
B. Establishing a type of deficiency (vitamin B
12
and/or
folic acid)
1) Low serum B
12
(in megaloblastic anemia or neuropathy
caused by B
12
deficiency).
2) Low serum and red cell folate (in megaloblastic anemia
caused by folate deficiency).
3) Homocysteine and methylmalonic acid level increased in
B
12
deficiency while homocysteine level increased in
folate deficiency.
C. Establishing a cause of deficiency.
Treatment
In vitamin B
12
deficiency, Hydroxocobalamin is given
(intramuscular).
In folate deficiency, folic acid is given (oral). If large
doses of folic acid are given in B
l2
deficiency they cause a
hematological response but may aggravate the
neuropathy. They should therefore not be given alone
unless B
12
deficiency has been excluded.
Prophylactic Therapy
Prophylactic Vitamin B
12
therapy is given to patients who
have: 1) Total gastrectomy. 2) Ileal resection.
Prophylactic folic acid is given in:
1) Pregnancy (to prevent a neural tube defect occurrence)
2) Chronic dialysis.
3) Hemolytic anemias.
4) Chronic myelofibrosis.
Pernicious anemia
A severe lack of IF due to gastric atrophy (autoimmune
destruction of gastric parietal cells). The ratio of incidence
in men and women is approximately 1:1.6 and the peak
age of onset is 60 years. Pernicious anemia associations
(occur more commonly with):
1) Female. 2) Blue eyes.
3) Premature greying. 4) Northern Europeans
5) Familial. 6) Blood group A.
7) Other autoimmune diseases (e.g. vitiligo, thyroid
diseases, Addison disease).
8) An association with HLA - B8, - B12 and - BW15.
Diagnosis
Pernicious anemia usually suspected from the clinical
picture and the findings of megaloblastic anemia due to
B
12
deficiency, confirmed by:
1) Presence of circulating gastric auto antibodies (important):
Parietal cell antibody.
IF antibody (blocking and binding) found.
2) Gastric biopsy usually shows atrophy of all layers.
3) A lack of IF has been demonstrated by cobalamin
absorption studies (Schilling technique): the urinary
secretion (Schilling) test was carried out with an oral trace
dose of radioactive cyanocobalamin with or without oral
IF and a (flushing) intramuscular dose of
hydroxocobalamin or cyanocobalamin so that any labeled
cobalamin absorbed would appear in a 24 hour urine (this
test is no longer available in most countries).
4) Low or absent Hcl or pepsin (now rarely performed).
Other Hemolytic anemias
These are anemias which result from an increase in RBC
destruction (mean lifespan of the red cell reduced).
Classifications:
1) Location of hemolysis (intravascular or extravascular).
2) Location of defect causing the hemolysis (intrinsic or
extrinsic).
3) Mode of onset (inherited or acquired).
Extravascular hemolysis: it's a normal process, RBCs
phagocytized by reticuloendothelial system cells (in
marrow, liver and spleen) after a mean lifespan of 120

Unit 11: Hematopoietic and Lymphoid Systems
160
days; Hb broken into portoporphyrin (which is converted
to bilirubin and excreted by liver), iron (binds to
transferrin and returns to marrow erythroblasts) and
globin chains (broken down to amino acids which are
reutilized for general protein synthesis).
Intravascular hemolysis: it's abnormal process, RBCs
break down inside the vessels, free Hb binds to
haptoglobin and haemopexin or converted to
methaemalbumin. These proteins are cleared by the liver
where the haem is broken down to recover iron & produce
bilirubin. The excess free Hb is filtered by the glomerulus,
and free Hb will enter the urine (haemoglobinuria), as
iron is released, the renal tubules become loaded with
haemosiderin (haemosiderinuria).
Hereditary hemolytic anemias (usually caused by
intrinsic defect):
1) Membrane defects e.g. hereditary spherocytosis.
2) Metabolic defect e.g. G6PD deficiency.
3) Haemoglobin defects e.g. Hb S, Hb C.
Acquired hemolytic anemias (usually caused by an
extrinsic defect):
1) Immune:
Autoimmune e.g. autoimmune hemolytic anemia
(warm and cold type).
Alloimmune e.g. hemolytic transfusion reactions,
hemolytic disease of the newborn.
Drug associated e.g. penicillin, quinidine, methyldopa
2) Red cell fragmentation syndromes.
3) March haemoglobinaemia.
4) Infections (Malaria, meningococcal or pneumococcal
septicaemia).
5) Chemical & physical agents (Wilson's disease, lead,
spider venom, burn).
6) Paroxysmal nocturnal haemoglobinuria (PNH)
Clinical features of hemolytic anemias:
1) Anemia and pallor of the mucous membranes.
2) Mild fluctuating jaundice.
3) Splenomegaly.
4) Dark urine (excess urobilinogen).
5) Pigment gall stones (bilirubin).
6) Aplastic crisis may precipitated by infection with
parvovirus or less frequently by folate deficiency.
7) Ulcers around the ankle (e.g. sickle cell anemia).
Main features of hemolytic anemia:
A. Increased red cell destruction
1) Serum unconjugated bilirubin increased:
Jaundice
Increased risk of gallstones.
2) Increased urine urinobilinogen and faecal
stercobilinogen.
3) Decreased serum haptoglobin and haemopexin.
4) Extravascular changes:
Splenomegaly.
Increased iron stores.
5) Intravascular changes:
Haemoglobinaemia (free Hb circulates in blood) and
haemoglobinuria (free Hb in urine).
Haemosiderinuria (haemosiderin in the urine, sign of
filtration of Hb through the kidney).
Methaemalbuminaemia (free Hb bound to albumin in blood).
Decreased iron stores.
B. Increased red cell production
1) Marrow expansion (bone changes)
2) Erythroid hyperplasia (decrease myeloid/erythroid ratio)
3) Reticulocytosis and polychromasia.
4) Increased folate requirements (macrocytosis).
C. Damaged red cells:
1) Morphology (e.g. microspherocytes, elliptocytes, fragments)
2) Osmotic fragility, autohaemolysis.
3) Short red cell survival and sites of destruction was
shown by 51Cr labelling study.
Hereditary spherocytosis
The inheritance is autosomal dominant. Hereditary
spherocytosis is usually extravascular hemolysis caused
by defects in the proteins of the red cell membranes. The
marrow produces red cells of normal biconcave shape but
these lose membrane and become increasingly spherical
as they circulate through the spleen. Ultimately, the
spherocytes are unable to pass through the splenic
microcirculation where they die prematurely. The
principal form of treatment is splenectomy.
Clinical features:
1) Anemia (can present at any age).
2) Fluctuating jaundice.
3) Splenomegaly.
4) Pigment gallstones.
5) Aplastic crises, usually precipitated by parvovirus
infection (increase in severity of anemia with low
reticulocytes count).
Laboratory findings:
1) Low Hb.
2) Reticulocytosis (reticulocytes count increased 5-20%).
3) Blood film show microspherocytes (smaller diameters
than normal red cells with no central pallor).

Unit 11: Hematopoietic and Lymphoid Systems
161
4) Osmotic fragility test is increased.
5) Autohaemolysis is increased & corrected by glucose.
6) The direct antiglobulin (Coomb's) test is normal.
GIucose 6 phosphate dehydrogenase
deficiency (G6PD):
The inheritance is sex-linked, affecting males and carried
by females. G6PD functions to reduce NADP while
oxidizing glucose 6 phosphate. NADP is needed for the
production of reduced glutathione. G6PD deficiency
renders the red cell susceptible to oxidant stress
(infection, acute illness, drugs, fava beans). G6PD
deficiency is usually intravascular hemolysis.
Clinical features:
G6PD deficiency is usually asymptomatic, the main
syndromes that occur are as follows:
1) Acute haemolytic anemia in response to oxidant stress
(rapidly developing intravascular hemolysis with
fever, chills, low back pain and haemoglobinuria). The
anemia may be self-limiting as new young red cells
are made with near normal enzyme levels.
2) Neonatal jaundice.
3) Congenital non spherocytic hemolytic anemia (rare).
Laboratory findings:
Between crises
1) The complete blood picture and film are normal.
2) The G6PD enzyme deficient.
During crisis
1) Low Hb
2) High reticulocytes count
3) Blood film may show:
Contracted and fragmented cells (bite & blister cells)
which have had Heinz bodies removed by the spleen.
Heinz bodies (oxidized, denatured haemoglobin)
may be seen in the reticulocyte preparation.
4) Intravascular hemolysis features.
5) The G6PD enzyme assay may give a false normal
level (higher enzyme level in young red cells).
Treatment:
1) Remove oxidant stress (stop drug, treat infection).
2) A high urine output is maintained.
3) Blood transfusion (for severe anemia).
4) Phototherapy & exchange transfusion (in neonatal jaundice)
Immune haemolytic anemias
They are characterized by a positive direct antiglobulin
test (DAT) also known as the Coombs' test.
A. Autoimmune haemolytic anemias
Autoimmune haemolytic anemias are caused by antibody
production by the body against its own red & divided into:
1) Warm autoimmune haemolytic anemias:
Either idiopathic or secondary to SLE, CLL,
lymphomas and drugs (e.g. methyldopa).
The Ab is usually IgG and binds to red cells best at 37°C
Extra vascular hemolysis generally.
Blood film show prominent spherocytosis.
The DAT is positive.
2) Cold autoimmune haemolytic anemias
Either idiopathic or secondary to infections (Mycoplasma
pneumonia, infectious mononucleosis) or lymphoma.
The Ab is usually IgM and binds to red cells best at 4°C.
Both intravascular & extravascular hemolysis can occur
Blood film show red cells agglutinate & less spherocytosis.
The DAT is positive.
B. Alloimmune hemolytic anemias
Antibody produced by one individual reacts with red cells
of another. Two important situations:
1) Hemolytic transfusion reactions (transfusion of ABO
incompatible blood).
2) Hemolytic disease of the newborn (when the
molecules produced by the mother pass through the
Red cell fragmentation syndromes
These arise through physical damage to red cells either on
abnormal surfaces (e.g. artificial heart valves or arterial
grafts) or as a microangiopathic hemolytic anemia (red
cells passing through abnormal small vessels (e.g.
disseminated intravascular coagulation DIC or thrombotic
thrombocytopenic purpura (TTP). The peripheral blood
contains many deeply staining red cell fragments.
March haemoglobinuria
This is caused by damage to red cells between the small
bones of the feet, usually during prolonged marching or
running. The blood film does not show fragments.
Paroxysmal nocturnal haemoglobinuria (PNH)
PNH is a rare, acquired, clonal disorder of marrow stem
cells in which there is deficient synthesis of the GPI
anchor, a structure that attaches several surface proteins to
the cell membrane (such as CD55 and CD59). The net
result is that CD55 and CD59 are absent from the cell
surface of all the cells derived from the abnormal stem
cell and render red cells sensitive to lysis by complement.
The main clinical problem in PNH is chronic

Unit 11: Hematopoietic and Lymphoid Systems
162
intravascular hemolysis, thrombosis and pancytopenia.
PNH may be diagnosed by flowcytometry and Ham's test
(red cell lysis in serum at low pH).
Genetic disorders of haemoglobin
Thalassaemia: which is a group of inherited disorders of
haemoglobin synthesis characterized by a reduced or
absent one or more of the globin chains of adult
haemoglobin e.g.
thalassaemia and
thalassaemia.
Haemoglobin variant: which is a group of inherited
disorders of haemoglobin synthesis characterized by
abnormal synthesis of globin chains of adult
haemoglobin e.g. Hb S, Hb C.
Normal blood Hb:
1) Hb A - α2 β2 chains (represent > 95%).
2) Hb F - α2 γ2 chains (represent < 1%).
3) Hb A
2
- chains (represent 1.5 - 3.5%).
α Thalassaemia
α Thalassaemia usually caused by gene deletions.
Normally, there are four copies of the α globin gene
(
/
), the clinical severity can be classified according
to the number of genes that are missing or inactive.
A deficiency of α chains leads to the production of excess γ
or β chains, which form Hb Bart’ s (γ4) and Hb H (β4).
These soluble tetramers do not precipitate extensively in the
bone marrow and hence erythropoiesis is more effective
than in β thalassaemia. However, Hb H is unstable and
precipitates in red cells as they age. The inclusion bodies
cause red cell membrane damage & obstruction in the
spleen leading to shortened red cell survival.
Thalassaemia trait
The a thalassaemia traits are caused by loss of one (-
/
) (silent carrier) or two a genes (-
/-
or --/
)
(thalassaemia minor).
Usually not associated with anemia.
MCV and MCH are low.
Red cell count is over 5.5 x 10
12
/L.
Haemoglobin electrophoresis is normal.
a/β globin chain synthesis studies or DNA analyses are
needed to diagnosis.
Hb H disease
Hb H disease is caused by loss of three a genes (--/-
).
Hb H contains 4β globin chains.
Moderately severe anemia (Hb 7-11 g/dL).
Splenomegaly.
MCV and MCH are low.
Reticulocyte preparations show Hb H (golf ball appearance)
Haemoglobin electrophoresis is abnormal (Hb H).
Hydrops fetalis
Hydrops fetalis is caused by loss of four a genes (--/--)
a chain is essential in fetal as well as in adult Hb, Loss
of all 4 genes completely suppresses a chain synthesis,
it is incompatible with life and leads to death in utero.
Hb Barts contains 4γ globin chains.
β Thalassaemia
β Thalassaemia caused by point mutations rather than
gene deletions in majority of cases. The β thalassaemia trait
either no β chain (β
o
) or small amounts (β
+
) are synthesized.
Excess a chains precipitate in erythroblasts and in mature
red cells causing severe ineffective erythropoiesis and
hemolysis. The greater the α chain excess, the more severe
the anemia. Production of γ chains helps to 'mop up' excess
a chains and to ameliorate the condition.
β Thalassaemia major (Cooley Anemia)
It is often a result of inheritance of two different
mutations: either homozygous β
o
(β
o
β
o
) or β
+
(β
+
β
+
)
or
heterozygotes (β
o
β
+
) or compound heterozygotes with
other Hb variants.
Clinical features
1) Severe anemia becomes apparent at 3-6 months after
birth (when the switch from γ to β chain production
occur), anemia is transfusion dependent.
2) Hepato-splenomegaly occurs as a result of excessive
red cell destruction, extramedullary haemopoiesis and
later because of iron overload.
3) Expansion of bones caused by intense marrow
hyperplasia leads to bone deformities.
4) Iron overload caused by repeated transfusions (unless
chelation therapy initiated). Iron damages the liver,
heart, endocrine organs.
5) Infections.
Laboratory Features
1) Severe anemia (low Hb).
2) MCV and MCH are low.
3) Reticulocytes count Increased.
4) Blood film show hypochromic, microcytic RCs, many
nucleated red cells, target cells & basophilic stippling.
5) Hb electrophoresis reveals absence or very low of Hb
A, high Hb F and Hb A
2
percentage is normal, low or
slightly raised.

Unit 11: Hematopoietic and Lymphoid Systems
163
6) a/β globin chain synthesis studies show an increased a:
β ratio with reduced or absent β chain synthesis.
7) DNA analysis is used to identify the defect.
Treatment
1) Regular blood transfusions are needed to maintain
the Hb over 10 g/dL at all times.
2) Regular folic acid.
3) Iron chelation therapy is used to treat iron overload
e.g. Desferrioxamine, Deferiprone and Deferasirox.
4) Splenectomy may be needed to reduce blood requirements.
Thalassaemia intermedia
Thalassaemia intermedia is a term used to define a group
of patients with
β
thalassaemia in whom the clinical
severity of the disease is spectrum range from mild
symptoms of the β thalassaemia trait to the severe
manifestations of β thalassaemia major. The diagnosis is a
clinical one that is based on the patient maintaining a
satisfactory Hb level of at least 6-7 g/dL at the time of
diagnosis without the need for regular blood transfusions
(not transfusion dependent). This is a clinical syndrome
which may be caused by a variety of genetic defects.
β Thalassaemia minor
β Thalassaemia trait is ββ
+
or ββ
o
.
Asymptomatic.
Mild anemia (Hb 10-12 g/dL).
MCV and MCH very low.
High red cell count (> 5.5 x 10
12
/L).
Haemoglobin electrophoresis show raised Hb A
2
(> 3.5%)
If both parents carry β thalassaemia trait, there is a 25%
risk of a thalassaemia major child. Therefore, prenatal
genetic counseling is important to patients with a partner
who also has a significant Hb disorder.
Haemoglobin S (Hb S)
Hb S is inherited Hb variant, the abnormality is caused by
substitution of valine for glutamic acid in position 6 in the
β globin chain (β change to S).
Hb S is insoluble and forms crystals when exposed to low
oxygen tension (e.g. hypoxia, dehydration, infections, cold
exposure) & the red cells sickle & may block different areas
of the microcirculation causing infarcts of various organs.
Sickle cell anemia (homozygous, Hb SS)
Clinical features
1) Severe haemolytic anemia punctuated by crises. The
symptoms of anemia are mild in relation to the
severity of the anemia because Hb S gives up oxygen
to tissues relatively easily compared with Hb A.
2) Painful vaso-occlusive crises (infarcts can occur in a
variety of organs including the bones, lung, kidney and
the spleen).
3) Visceral sequestration crises (sickling within organs &
pooling of blood) with a severe exacerbation of anemia
4) Aplastic crises occur as a result of infection with
parvovirus or from folate deficiency (sudden fall in Hb
and reticulocytes count).
5) Haemolytic crises (increased rate of haemolysis with a
fall in Hb but rise in reticulocytes count).
6) Other clinical features: ulcers of the lower legs are
common. The spleen is enlarged in infancy and early
childhood but later is often reduced in size as a result
of infarcts (autosplenectomy). Pulmonary
hypertension, a proliferative retinopathy and priapism.
Laboratory findings
Hb is usually 6-9 g/dL.
Blood film show sickle cells, target cells, Howell Jolly
bodies.
Screening tests for sickling are positive when the
blood is deoxygenated (e.g. sodium metabisulphite).
Haemoglobin electrophoresis: Hb S is high, no Hb A
is detected and Hb F is variable amount (5-15%).
Treatment
Avoid factors known to precipitate crises e.g.
dehydration, anoxia, infections and cold exposure.
Regular folic acid.
Crises: treat by rest, warmth, rehydration, antibiotics,
analgesia, exchange transfusion (aim to reduce Hb S
less than 30%).
Blood transfusion (given only in severe anemia with
symptoms).
Hydroxyurea can increase Hb F.
Sickle cell trait (Heterozygous, Hb βS)
Benign condition with no anemia and normal
appearance of red cells on a blood film (unless
exposed to low oxygen tension).
Impaired urine concentrating ability and haematuria
can occur (due to minor infarcts of the renal papillae).
Haemoglobin electrophoresis: The ratio of Hb A to Hb
S is 60 : 40%

Unit 11: Hematopoietic and Lymphoid Systems
164
WHITE CELL DISORDERS
Acute leukaemia
Genetic damage is believed to involve several key
biochemical steps resulting in: an increased rate of
proliferation; reduced apoptosis and a block in cellular
differentiation. Together these events cause accumulation
of the early bone marrow haemopoietic cells (blast cells).
Acute leukaemia is defined as the presence of over 20%
of blast cells in the blood or bone marrow at clinical
presentation. It can be diagnosed with even less than 20%
blasts if specific leukaemia associated cytogenetic or
molecular genetic abnormalities are present, types:
1) Acute myeloid leukaemia (AML).
2) Acute lymphoblastic leukaemia (ALL).
The aetiology of leukemia:
1) Inherited factors: e.g. Down's syndrome, Fanconi's
anemia.
2) Environmental influences:
Chemicals: chronic exposure to benzene or industrial
solvents may cause AML.
Chemotherapy: alkylating agents and melphalan
predispose to AML.
Ionizing radiation.
Infection: HTLV-1 cause Adult T-cell leukemia
Lymphoma.
3) Preleukemic syndrome: e.g. myelodysplastic syndrome,
myeloproliferative neoplasm.
Acute lymphoblastic leukaemia (ALL)
ALL is the most common malignancy of childhood (highest
at 3-7 years), a secondary rise after the age of 40 years.
The common precursor B type (CDI0+) is the most usual
in children. has an equal sex incidence but there is a male
predominance for T-cell ALL (T-ALL).
Classification
is usually based on the:
1) Morphology: French American British (FAB) classification:
The L1 type show uniform, small blast cells with scanty
cytoplasm.
The L2 type comprise larger blast cells with more
prominent nucleoli & cytoplasm & with more heterogeneity
The L3 blasts are large with prominent nucleoli, strongly
basophilic cytoplasm and cytoplasmic vacuoles.
2) Immunophenotype: Immunological markers can be used to
divide ALL cases into early pre-B, pre-B, B& T-cell subtypes
3) Chromosomes and genetic analysis:
Hyperdiploid (cells have >50 chromosomes) or hypodiploid.
t(12; 21) TEL-AMLl translocation.
t(9; 22) Philadelphia chromosome.
llq23 Translocations (MLL gene).
Clinical features
1) Bone marrow failure:
Anemia (fatigue, pallor).
Neutropenia (infections).
Thrombocytopenia (spontaneous bruises, bleeding).
2) Organ infiltration:
Bone pain.
Lymphadenopathy, splenomegaly (moderate) and
hepatomegaly.
Meningeal syndrome (headache, nausea and vomiting,
blurring of vision and diplopia).
Testicular swelling or signs of meditational
compression in T-ALL.
Acute myeloid leukaemia (AML)
It is the common form of acute leukaemia in adults and is
increasingly common with age. AML forms only a minor
fraction (10-15%) of the leukaemias in childhood.
Classification
is usually based on the:
1) Morphology (FAB criteria):
AML M0 undifferentiated form.
AML M1 without maturation.
AML M2 with granulocytic maturation.
AML M3 acute promyelocytic.
AML M4 granulocytic and monocytic maturation.
AML M5 monoblastic (M5a) or monocytic (M5b).
AML M6 erythroleukaemia.
AML M7 megakaryoblastic.
2) Immunophenotype: CDI3, CD33, CD117, Glycophorin
(M6), platelet antigens (e.g. CD41) in (M7) and
myeloperoxidase in (M0).
3) Chromosomes and genetic analysis:
Normal karyotype.
t(8; 21), t(15; 17) and inv(16).
Nucleophosmin (NPM) gene mutations
Internal tandem repeats of the FLT-3 gene.
Clinical features
1) Bone marrow failure:
Anemia (fatigue, pallor).
Neutropenia (infections).
Thrombocytopenia (spontaneous bruises, bleeding).

Unit 11: Hematopoietic and Lymphoid Systems
165
2) Organ infiltration:
Gum hypertrophy, skin involvement and CNS disease
are characteristic of the AML M4 and M5.
Granulocytic sarcoma (isolated mass of leukaemic blasts)
3) Disseminated intravascular coagulation (DIC) is
characteristic of the AML M3.
Laboratory findings:
1) Complete blood picture:
Normochromic, normocytic anemia.
Thrombocytopenia.
The total white cell count may be decreased, normal or
increased to 200 x 10
9
/l or more.
2) Blood film shows a variable numbers of blast cells.
3) The bone marrow hypercellular with >20% leukaemic
blasts.
4) CSF examination may show leukaemic cells.
5) Coagulation tests for DIC are often positive in patients
with AML M3.
6) Biochemical tests: serum uric acid and serum lactate
dehydrogenase increased.
7) Radiography: may reveal lytic bone lesions or a
meditational mass (enlargement of the thymus and/or
lymph nodes characteristic of T-ALL).
Differentiation of ALL from AML
1) Morphology: lymphoblast vs. myeloblast (Auer rods).
2) Cytochemistry (special stains):
Myeloperoxidase and Sudan black are positive in
AML (including Auer rods) but negative in ALL.
Nonspecific esterase is positive in AML M4 and M5
but negative in ALL.
Periodic acid Schiff is positive in ALL (coarse block),
also positive in AML M6 (fine blocks).
Acid phosphatase is positive in T-ALL (Golgi
staining), also positive in AML M6 (diffuse).
3) Immunoglobulin and TCR genes:
Precursor B-ALL show clonal rearrangement of
immunoglobulin genes.
T-ALL show clonal rearrangement of TCR genes.
AML show germline configuration of
Immunoglobulin and TCR genes.
4) Immunophenotype:
AML show positive CDI3 and CD33.
B ALL show positive CD19, cCD22 and TdT.
T ALL show positive cCD3, CD7 and TdT.
5) Chromosomes and genetic analysis
Treatment
General supportive therapy for bone marrow failure
includes blood product support, treat infections, DIC and
prevention of tumour lysis syndrome.
Chemotherapy and sometimes radiotherapy (different
protocols according to age), several phases:
1) Remission induction: the aim is to rapidly kill most
of the tumour cells and get the patient into remission
(less than 5% blasts in the bone marrow).
2) Consolidation (Intensification): two or three courses
use high doses of multidrug chemotherapy in order to
completely reduce or eliminate the tumour burden to
very low levels.
3) CNS prophylaxis: to prevent or treat central nervous
system disease (only for ALL).
4) Maintenance therapy for 2 to 3 years (for ALL and
AML M3 only).
Stem cell transplantation reduces the rate of relapse but
adds further toxicity to the treatment.
All-trans retinoic acid (ATRA) therapy is given in
conjunction with chemotherapy for the M3 disease.
Poor prognostic factor in ALL:
1) High WBC at presentation (e.g. >50 x 10
9
/L).
2) Boys.
3) T-ALL Immunophenotype in children.
4) Age: adult or infant <2 years.
5) Cytogenetics: Ph+, llq23 rearrangements.
6) Time to clear blasts from blood (>1 week)
7) Time to remission (>4 weeks)
8) CNS disease at presentation
9) Minimal residual disease still positive at 3-6 months.
Prognostic factor in AML:
1) Cytogenetics: t(15; 17), t(8; 21), inv(16) and NPM
mutation carry better prognosis than other
chromosomes and genetic lesion.
2) Bone marrow response to remission induction: <5%
blasts after first course show better prognosis.
3) Age of patient: <60 years show better prognosis than >
60 years.
4) Onset: primary show better prognosis than secondary.

Unit 11: Hematopoietic and Lymphoid Systems
166
Chronic Leukaemia
The chronic leukaemias have slower progression.
Paradoxically, they are also more difficult to cure.
Chronic leukaemias can be subdivided into:
Chronic myeloid leukaemia represent about
15% of all leukaemias.
25% of all leukaemias.
Chronic myeloid leukaemia (CML)
Chronic myeloid leukaemia (CML) is a clonal disease that
results from an acquired genetic change in a stem cell.
This altered stem cell proliferates and generates a
population of differentiated cells that gradually displaces
normal haemopoiesis and leads to a greatly expanded total
myeloid mass. CML has three phases: chronic phase,
accelerated phase and blastic crisis.
Leukaemogenesis
Philadelphia chromosome is an acquired cytogenetic
anomaly that is characterizes in all leukaemic cells in CML.
90-95% of CML patients have Ph chromosome.
Reciprocal translocation of chromosome 22 and
chromosome 9; t(9;22).
BCR-ABL1 fusion gene generate a fusion protein that has
tyrosine kinase activity in excess of the normal. Growth
and survival of hematopoietic cells increased and become
independent of cytokines.
Clinical features
Chronic phase
The median age of onset is 50 - 60 years. The incidence is
slightly higher in males than in females.
Asymptomatic, 50% of CML can be discovered in a
routine blood examination.
CML patients may present with:
Anemia (fatigue, shortness of breath).
Haemorrhage (bruising or bleeding).
Abdominal discomfort (splenomegaly).
Weight loss.
Excessive sweating.
Less frequent symptoms:
Splenic infarction (left upper-quadrant and left
shoulder pain).
Leucostasis features (very high leucocyte counts) e.g.
headache, blurred vision, cerebrovascular accidents
and dyspnea.
Visual disturbances (retinal haemorrhages).
Gout.
Blastic transformation
Spleen is frequently enlarged and may be painful.
Liver may become very large.
Fever and lymphadenopathy (rare in chronic phase).
Lytic lesions of bone (rare).
Laboratory findings:
Chronic phase
1) Hb low (normochromic, normocytic anemia).
2) WBCs count is high (20 – 200 × 10
9
/L).
3) Platelet numbers are usually increased, normal or even reduced.
4) Blood film shows:
A full spectrum of granulocyte series cells, ranging
from blast forms to mature neutrophils (myelocytes
and neutrophils predominating).
Blast less than 12%.
The percentages of eosinophils and basophils are
usually increased.
5) Bone marrow aspirate shows:
Hypercellular fragments and trails.
A full spectrum of granulocyte series cells (resembling
that of CML blood).
Blast cells in chronic phase 2 – 10%.
Eosinophils and basophils are usually prominent.
Megakaryocytes are very numerous.
6) Bone marrow biopsy shows:
Hypercellularity (complete loss of fat spaces).
The reticulin content (fibrosis) may be normal or increased.
7) Neutrophils alkaline phosphatase score is invariably low
(increase in infections).
8) BCR – ABL1 transcripts can be demonstrated in the blood
(diagnostic) using real time quantitative polymerase chain
reaction (RQ - PCR).
9) Serum uric acid is usually normal or slightly raised.
Lactate dehydrogenase is usually raised.
10) The serum vitamin B
12
and B
12
binding capacity are
greatly increased.
Accelerated phase:
1) Anemia in the presence of a normal leucocyte count.
2) Platelets number may be greatly increased (>1000 ×
10
9
/L) or reduced (< 100 × 10
9
/L) in a manner not
accounted for by treatment.
3) Basophilia ≥ 20%.
4) The marrow shows increased numbers of blast cells (10 -
19 %) or fibrosis.
5) Serum uric acid increased
Blastic crisis: Blastic crisis is defined by the presence
of more than 20% blasts in the blood or marrow.

Unit 11: Hematopoietic and Lymphoid Systems
167
Treatment
1) Tyrosine kinase inhibitor: Imatinib mesylate (Glivec,
Gleevec).
2) Chemotherapy: Hydroxyurea, Busulfan.
3) Interferon Alfa.
4) Allogeneic stem cell transplant.
Course and prognosis
CML usually shows an excellent response to imatinib in
the chronic phase.
If death occurs it is usually from terminal acute
transformation or from intercurrent haemorrhage or
infection.
Chronic lymphocytic leukaemia (CLL)
Chronic lymphocytic leukaemia (CLL) is the most
common form of leukemia in adults over the age of 50
years. The tumour cell appears to be a relatively mature B
cell with weak surface expression of IgM or IgD. The
cells accumulate in the blood, bone marrow, liver, spleen
and lymph nodes as a result of a prolonged lifespan with
impaired apoptosis.
Cytogenetics:
The most common chromosomes abnormalities are:
Deletion of 13q14.
Trisomy 12.
Deletions at llq23.
Structural abnormalities of 17p involving the p53 gene
Clinical features
1) Peak incidence between 60 and 80 years of age. The male
to female ratio is 2 : 1.
2) Most cases are diagnosed when a routine blood test is
performed.
3) Symmetrical enlargement of cervical, axillary or inguinal
lymph nodes is the most frequent clinical sign. The nodes
are usually discrete and non tender.
4) Anemia may be present.
5) Thrombocytopenia may be present (bruising or purpura).
6) Splenomegaly and, less commonly, hepatomegaly are
common in later stages.
7) Infection (bacterial, viral or fungal) due to immune
suppression (resulting from hypogammaglobulinaemia
and cellular immune dysfunction).
Laboratory findings
1) Lymphocytosis. The absolute lymphocyte count is >5 x
10
9
/L and may be up to 300 X 10
9
/L or more.
2) Blood film shows small mature lymphocytes, smudge or
smear cells.
3) Immunophenotyping of the lymphocytes shows:
CD19+ (B cells).
Weakly expressing of surface IgM or IgD.
Expression of only one form of light chain, either κ or
λ (monoclonal).
CD5+ and CD23+ but CD79b - and FMC7- (characteristic).
4) Normocytic normochromic anaemia is present in later
stages as a result of marrow infiltration, hypersplenism or
autoimmune haemolysis.
5) Thrombocytopenia occurs in later stages.
6) Bone marrow aspiration shows lymphocytic replacement
of normal marrow elements. Lymphocytes comprise 25-
95% of all the cells.
7) Bone marrow biopsy reveals nodular, diffuse or
interstitial involvement by lymphocytes.
8) Reduced concentrations of serum Ig.
9) Autoimmunity directed against cells of the haemopoietic
system is common. Autoimmune haemolytic anaemia is
most frequent but immune thrombocytopenia, neutropenia
and red cell aplasia are also seen.
Good Prognostic markers:
1) Stage: Binet A (Rai 0-1).
2) Female.
3) Slow lymphocyte doubling time.
4) Nodular infiltration in bone marrow biopsy.
5) Cytogenetics: Deletion 13q14.
6) Ig genes mutation status: hypermutated (unmutated Ig
genes has an unfavorable prognosis).
7) Negative CD38 expression.
8) Low ZAP-70 expression.
9) Low CLLU.l expression.
10) Normal LDH.
Treatment:
1) Chemotherapy: alkylating agent or Purine analogues.
2) Monoclonal antibodies: Campath-1H (anti-CD52),
Rituximab (anti CD20).
3) Others: corticosteroids, radiotherapy, splenectomy, Ig replacement.
4) Allogeneic stem cell transplantation.

Unit 11: Hematopoietic and Lymphoid Systems
168
Course of disease
Many patients in Binet stage A or Rai stage 0 or I never
need therapy.
For those who do need treatment, typical pattern is that of
a disease that is responsive to several courses of
chemotherapy before the gradual onset of extensive bone
marrow infiltration, bulky disease and recurrent infection.
The disease may transform into a high grade lymphoma
(Richter's transformation) or there may be the appearance
of an increasing number of prolymphocytes that are
resistant to treatment.