
Unit 5: Neoplasia
77
Nomenclature
Benign Tumors
Malignant Tumors

Unit 5: Neoplasia
78

Unit 5: Neoplasia
79
Characteristics of benign andmalignant neoplasms
1) Malignant transformation (Differentiation and Anaplasia)

Unit 5: Neoplasia
80
Dysplasia

Unit 5: Neoplasia
81
2) Rate of Growth (Growth rate of transformed cells)

Unit 5: Neoplasia
82
3) Local Invasion
4) Metastasis

Unit 5: Neoplasia
83
Epidemiology of neoplasm
Cancer epidemiology can contribute substantially to
knowledge about the origin of cancer. Now well-
established concept that cigarette smoking is causally
associated with lung cancer arose primarily from
epidemiologic studies. Epidemiology of cancer is focused
on several subjects which are:
Cancer incidence
Incidence of cancers all over the world will give idea
about the effect of prevention and treatment cancers.
Over several decades, the death rates of many forms of
malignant neoplasia have changed. Particularly notable is
the significant increase in the overall cancer death rate
among men that was attributable largely to lung cancer, but
this has finally begun to drop. In contrast, the overall death
rate among women has fallen slightly, mostly as a result of
the decline in death rates from cancers of the uterine cervix,
stomach, and large bowel. These welcome trends have
more than counterbalanced the striking climb in the rate of
lung cancer among women, which not long ago was a
relatively uncommon form of neoplasia in this sex. The
declining death rate from cervical cancer is directly related
to widespread use of cytologic smear studies for early
detection of this tumor while it is still curable. The causes
of decline in death rates for cancers of the stomach are
obscure; however, there have been speculations about
decreasing exposure to dietary carcinogens.
Geographical and environmental factors
The incidences of certain cancer are different from on
area to other in our world this may be due to certain
environmental or genetic factors that may occur due to
certain environmental factors.
This notion is supported by the geographic differences in
death rates from specific forms of cancer. For example,
death rates from breast cancer are about fourfold to
fivefold higher in the United States and Europe compared
with Japan. Conversely, the death rate for stomach
carcinoma in men and women is about seven times higher
in Japan than in the United States. Liver cell carcinoma is
relatively infrequent in the United States but is the most
lethal cancer among many African populations. Nearly all
the evidence indicates that these geographic differences
are environmental rather than genetic in origin.
Age
In general, the frequency of cancer increases with age.
Most cancer mortality occurs between ages 55 and 75; the
rate declines, along with the population base, after age 75.
The rising incidence with age may be explained by:
1) The accumulation of somatic mutations associated
with the emergence of malignant neoplasms.
2) The decline in immune competence that accompanies
aging also may be a factor.
Cancer causes slightly more than 10% of all deaths among
children younger than 15years. The major lethal cancers in
children are leukemia, tumors of the central nervous
system, lymphomas, soft tissue sarcomas & bone sarcomas.
Heredity
The evidence now indicates that for many types of cancer,
including the most common forms, there exist not only
environmental influences but also hereditary
predispositions. Hereditary forms of cancer can be
divided into three categories
A- Inherited cancer syndromes: (Autosomal
dominant pattern)
Inherited cancer syndromes include several well-defined
cancers in which inheritance of a single mutant gene
greatly increases the risk of developing a tumor. The
predisposition to these tumors shows an autosomal
dominant pattern of inheritance. Childhood
retinoblastoma is the most striking example of this
category.
Tumors within this group often are associated with a
specific marker phenotype. There may be multiple benign
tumors in the affected tissue, as occurs in familial polyposis
of the colon and in multiple endocrine neoplasia.
B – Familial cancers
Virtually all the common types of cancers that occur
sporadically have been reported to occur in familial
forms. Examples include carcinomas of colon, breast,
ovary, and brain. Features that characterize familial
cancers include early age at onset, tumors arising in two
or more close relatives of the index case, and sometimes
multiple or bilateral tumors. Familial cancers are not
associated with specific marker phenotypes. For example,
in contrast to the familial adenomatous polyposis
syndrome, familial colonic cancers do not arise in
preexisting benign polyps. The transmission pattern of
familial cancers is not clear.
Unit 5: Neoplasia
84
C- Autosomal recessive syndromes (Defective
DNA repair)
Besides the dominantly inherited precancerous conditions,
a small group of autosomal recessive disorders is
collectively characterized by chromosomal or DNA
instability. One of the best-studied examples are
xeroderma pigmentosum, Fanconi anemia, ataxic
talangectsia. in which DNA repair is defective.
Acquired Preneoplastic Disorders
In addition to the genetic influences described earlier,
certain clinical conditions are well-recognized
predispositions to the development of malignant neoplasia
and are referred to as preneoplastic disorders. This
designation is unfortunate because it implies a certain
inevitability, but in fact, although such conditions may
increase the likelihood, in most instances cancer does not
develop. A brief listing of the chief conditions follows:
* Persistent regenerative cell replication (e.g., squamous
cell carcinoma in the margins of a chronic skin fistula or
in a long-unhealed skin wound; hepatocellular carcinoma
in cirrhosis of the liver)Hyperplastic and dysplastic
proliferations (e.g., endometrial carcinoma in atypical
endometrial hyperplasia; bronchogenic carcinoma in the
dysplastic bronchial mucosa of habitual cigarette
smokers)Chronic atrophic gastritis (e.g., gastric
carcinoma in pernicious anemia or following long-
standing Helicobacter pylori infection)Chronic ulcerative
colitis (e.g., an increased incidence of colorectal
carcinoma in long-standing disease)Leukoplakia of the
oral cavity, vulva, or penis (e.g., increased risk of
squamous cell carcinoma)Villous adenomas of the colon
(e.g., high risk of transformation to colorectal carcinoma)
Carcinogenesis: the molecular basis
of cancer
Non-lethal genetic damage lies at the heart of
carcinogenesis. Such genetic damage (or mutation) may
be acquired by the action of environmental agents, such as
chemicals, radiation, or viruses, or it may be inherited in
the germ line. The genetic hypothesis of cancer implies
that a tumor mass results from the clonal expansion of a
single progenitor cell that has incurred genetic damage
(i.e., tumors are monoclonal).
Four classes of normal regulatory genes-growth-
promoting proto-oncogenes, growth-inhibiting tumor
suppressor genes, genes that regulate programmed cell
death (i.e., apoptosis), and genes involved in DNA repair-
are the principal targets of genetic damage. Collectively
the genetic alterations in tumor cells confer upon them
growth and survival advantages over normal cells.
Carcinogenesis is a multi-steps process resulting from the
accumulation of multiple mutations. Over a period of time
many tumors become more aggressive by acquiring
greater malignant potential .This phenomena is referred to
as tumor progression. Increasing malignancy is often
acquired step by step. At the molecular level, tumor
progression result from multiple mutations that
accumulate independently in different cells generating
subclones with different characteristics such as ability to
invade, rate of growth, metastatic ability, hormonal
responsiveness and susceptibility to anti neoplastic drugs.
Even though most malignant tumors are monoclonal in
origin by the time they become clinically evident their
constituent cells are extremely hetrogenous.
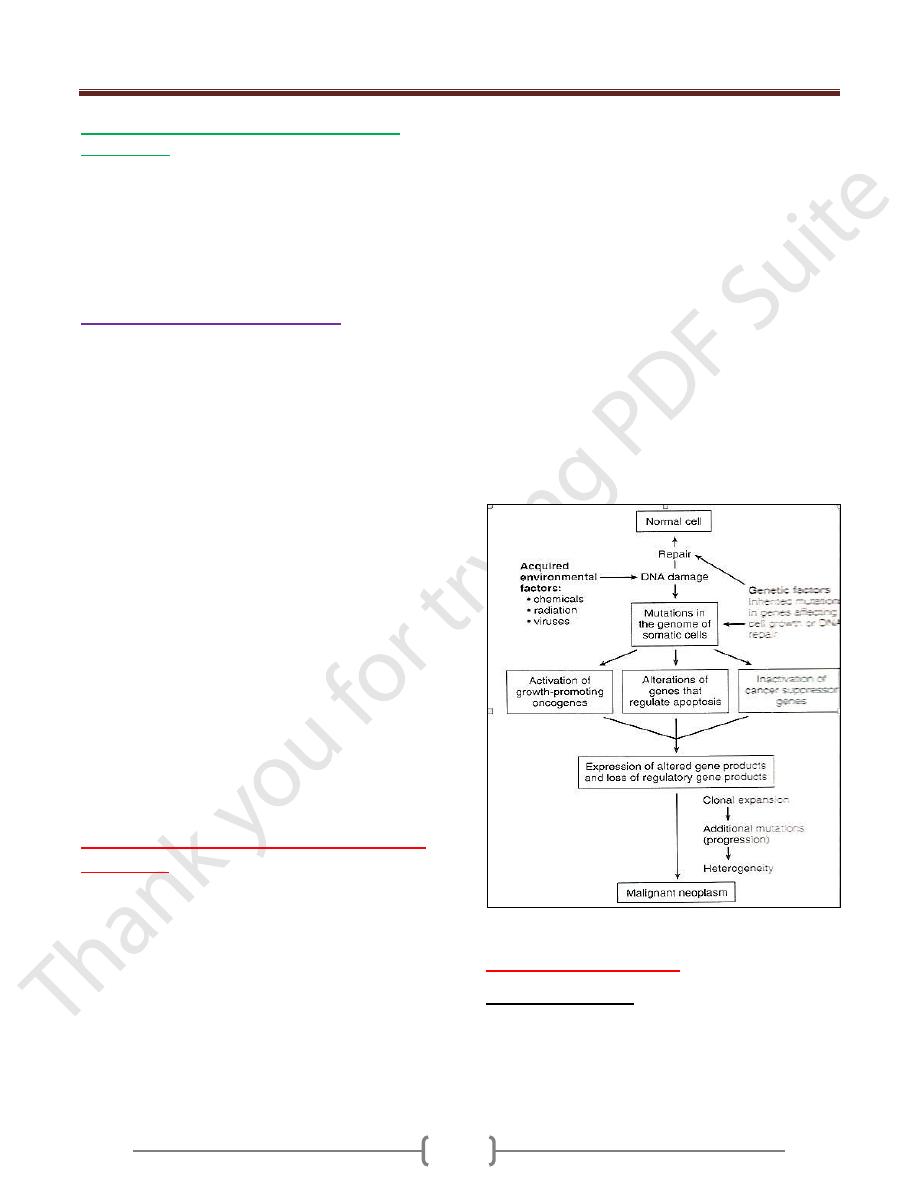
Illustration of Pathogenesis of cancer
Hallmarks of cancer
Cancer critical genes
These genes are had a role in seven fundamental changes in
cell physiology that together determine its malignant attributes
1) Self-sufficiency in growth signals
2) Insensitivity to growth-inhibitory signals
3) Evasion of apoptosis

Unit 5: Neoplasia
85
4) Limitless replicative potential (i.e., overcoming cellular
senescence and avoiding mitotic catastrophe)
5) Development of sustained angiogenesis
6) Ability to invade and metastasize
7) Genomic instability resulting from defects in DNA repair
Mutation in genes that regulate some or all of the above
cellular traits are seen in every cancer and hence these
will form the basis of the molecular origins of cancer.
1- Self – sufficiency in growth signals
Genes that promote autonomous cell growth in cancer cells
are called oncogenes. They are derived by mutations in
proto-oncogenes and are characterized by the ability to
promote cell growth in the absence of normal growth-
promoting signals. Their products, called oncoproteins,
resemble the normal products of proto-oncogenes except
that oncoproteins are devoid of important regulatory
elements & their production in the transformed cells does
not depend on growth factors or other external signals.
Under physiologic conditions, cell proliferation can be
readily resolved into the following steps:
1) The binding of a growth factor to its specific receptor on
the cell membrane
2) Transient and limited activation of the growth factor
receptor, which in turn activates several signal- transducing
proteins on the inner leaflet of the plasma membrane
3) Transmission of the transduced signal across the cytosol
to the nucleus via second messengers or a cascade of
signal transduction molecules
4) Induction and activation of nuclear regulatory factors that
initiate DNA transcription
5) Entry and progression of the cell into the cell cycle,
resulting ultimately in cell division.
Self – sufficiency involve the following
A – Growth factors
All normal cells require stimulation by growth factors to
undergo proliferation. Most soluble growth factors are
made by one cell type and act on a neighboring cell to
stimulate proliferation (paracrine action). Many cancer cells
acquire growth self-sufficiency, however, by acquiring the
ability to synthesize the same growth factors to which they
are responsive. For example, many glioblastomas secrete
platelet-derived growth factor (PDGF) and express the
PDGF receptor, and many sarcomas make both
transforming growth factor-α (TGF-α) and its receptor.
B- Growth factors receptors
Mutant genes present in two forms:
1) Formation of mutant receptor protein that deliver
continuous mitogenic signals to cells even in the absence
of the relevant growth factor in the environment.
2) Overexpression of growth factor receptors that render
cancer cells hyper-responsive to levels of growth factor that
would not normally trigger proliferation. As examples are
EGF (epidermal growth factor) overexpression in 80% of
squamous cell carcinoma. HER/NEU receptor is amplified
in 25% of breast carcinoma which indicated bad prognosis.
C- Single – transducing proteins
A relatively common mechanism by which cancer cells
acquire growth autonomy is mutations in genes that
encode various components of the signaling molecules
couple growth factor receptors to their nuclear targets.
Many such signaling proteins are associated with the
inner leaflet of the plasma membrane, where they receive
signals from activated growth factor receptors and
transmit them to the nucleus, either through second
messengers or through a cascade of phosphorylation and
activation of signal transduction molecules. Two
important members in this category are RAS and ABL.
D – Nuclear transcription factors:
Transcription is the synthesis of messenger RNA (mRNA)
from a DNA this is a key step in the formation of protein
coded by DNA. Transcription factors are proteins
necessary for RNA polymerase to initiate transcription of
mRNA molecule from its DNA template.
As example of this group (MYS) gene which showed
mutation by translocation between chromosomes (8 : 14)
which occur in burkitt lymphoma & also breast, colon, lung.
E – cyclins and cyclin – dependent Kinase (CDKs).
They are activated by binding to cyclins (so called because
of cyclic nature of their production and degradation). The
CDK- cyclin complexes activate the crucial RB protein that
derives the cell cycle. On completion of this task, cyclin
levels decline rapidly. Cyclin D, E , A , and B appear
sequentially during the cell cycle to one or more CDK.
Mutation that dsregulate the activity of cyclins and CDKs
would favor cell proliferation .The cyclin D genes are
overexpressed in many cancers including breast, esophagus,
melanoma, sarcom. While cyclins activate the CDKs their
inhibitors (CDKIs) such as (P21 ,P 27 and P57 ) silence the
CDKs and exert negative control the cell cycle. The CDKIs
are frequently mutated or otherwise silenced in many
malignancies. (Pancreatic, esophagus or glioblastoma.

Unit 5: Neoplasia
86
2) Insensitivity to Growth Inhibitory Signals
RB Gene: Governor of the Cell Cycle
P53 Gene: Guardian of the Genome

Unit 5: Neoplasia
87
Transforming Growth Factor-β Pathway (TGF-β)
Adenomatous polyposis Coli-β-Catenin Pathway
3) Avoidance of apoptosis

Unit 5: Neoplasia
88
4) Unlimited replicative potential
5) Development of sustain angiogenesis

Unit 5: Neoplasia
89
6) Invasion & Metastasis

Unit 5: Neoplasia
90

Unit 5: Neoplasia
91
Etiology of cancer: carcinogenic agents
Three classes of carcinogenic agents can be identified:
(1) chemicals, (2) radiant energy, and (3) microbial
agents. Chemicals and radiant energy are documented
causes of cancer in humans, and oncogenic viruses are
involved in the pathogenesis of tumors in several animal
models and at least in some human tumors. In the
following discussion, each class of agents is considered
separately, but it is important to note that several may act
in concert or sequentially to produce the multiple genetic
abnormalities characteristic of neoplastic cells.
Chemical Carcinogens
Direct-Acting Agents
Direct-acting agents require no metabolic conversion to
become carcinogenic. They are in general weak
carcinogens but are important because some of them are
cancer chemotherapeutic drugs (e.g., alkylating agents)
that have successfully cured, controlled, or delayed
recurrence of certain types of cancer (e.g., leukemia,
lymphoma, Hodgkin lymphoma, and ovarian carcinoma),
only to evoke later a second form of cancer, usually
leukemia. This situation is even more tragic when their
initial use has been for non-neoplastic disorders, such as
rheumatoid arthritis or Wegener granulomatosis. The risk
of induced cancer is low, but its existence dictates
judicious use of such agents.
Indirect-Acting Agents
The designation indirect-acting agent refers to chemicals
that require metabolic conversion to an ultimate
carcinogen before they become active. Some of the most
potent indirect chemical carcinogens-the polycyclic
hydrocarbons-are present in fossil fuels. For example,
benzo[a]pyrene and other carcinogens are formed in the
high-temperature combustion of tobacco in cigarette
smoking. These products are implicated in the causation
of lung cancer in cigarette smokers. Polycyclic
hydrocarbons may also be produced from animal fats
during the process of broiling meats and are present in
smoked meats & fish. The principal active products in
many hydrocarbons are epoxides, which form covalent
adducts (addition products) with molecules in the cell,
principally DNA, but also with RNA and proteins.
The aromatic amines and azo dyes are another class of
indirect-acting carcinogens. Before its carcinogenicity was
recognized, β-naphthylamine was responsible for a 50-fold
increased incidence of bladder cancers in heavily exposed
workers in the aniline dye and rubber industries. Many
other occupational carcinogens. Because indirect-acting
carcinogens require metabolic activation for their
conversion to DNA-damaging agents, much interest is
focused on the enzymatic pathways that are involved, such
as the cytochrome P-450-dependent monooxygenases. The
genes that encode these enzymes are polymorphic, and
enzyme activity varies among different individuals. It is
widely believed that the susceptibility to chemical
carcinogenesis depends at least in part on the specific allelic
form of the enzyme inherited. Thus, it may be possible in
the future to assess cancer risk in a given individual by
genetic analysis of such enzyme polymorphisms.
A few other agents merit brief mention. Aflatoxin B
1
is of
interest because it is a naturally occurring agent produced
by some strains of Aspergillus, a mold that grows on
improperly stored grains and nuts. There is a strong
correlation between the dietary level of this food
contaminant &the incidence of hepatocellular carcinoma in
some parts of Africa and the Far East. Additionally, vinyl
chloride, arsenic, nickel, chromium, insecticides, fungicides
& polychlorinated biphenyls are potential carcinogens in
the workplace and about the house. Finally, nitrites used as
food preservatives have caused concern, since they cause
nitrosylation of amines contained in the food. The
nitrosoamines so formed are suspected to be carcinogenic.
Mechanisms of Action of Chemical Carcinogens
Because malignant transformation results from mutations,
it should come as no surprise that most chemical
carcinogens are mutagenic. Indeed, all direct and ultimate
carcinogens contain highly reactive electrophile groups
that form chemical adducts with DNA, as well as with
proteins and RNA. Although any gene may be the target
of chemical carcinogens, the commonly mutated
oncogenes and tumor suppressors, such as RAS and p53,
are important targets of chemical carcinogens. Indeed,
specific chemical carcinogens, such as aflatoxin B
1
,
produce characteristic mutations in the p53 gene, such
that detection of the "signature mutation" within the p53
gene establishes aflatoxin as the causative agent. These
associations are proving useful tools in epidemiologic
studies of chemical carcinogenesis.
Carcinogenicity of some chemicals is augmented by
subsequent administration of promoters (e.g., phorbol
esters, hormones, phenols, and drugs) that by themselves
are nontumorigenic. To be effective, repeated or sustained
exposure to the promoter must follow the application of
the mutagenic chemical, or initiator. The initiation-
promotion sequence of chemical carcinogenesis raises an
important question: Since promoters are not mutagenic,

Unit 5: Neoplasia
92
how do they contribute to tumorigenesis? Although the
effects of tumor promoters are pleiotropic, induction of
cell proliferation is a sine qua non of tumor promotion. It
seems most likely that while the application of an initiator
may cause the mutational activation of an oncogene such
as RAS, subsequent application of promoters leads to
clonal expansion of initiated (mutated) cells. Forced to
proliferate, the initiated clone of cells accumulates
additional mutations, developing eventually into a
malignant tumor. Indeed, the concept that sustained cell
proliferation increases the risk of mutagenesis, and hence
neoplastic transformation, is also applicable to human
carcinogenesis. For example, pathologic hyperplasia of
the endometrium) and increased regenerative activity that
accompanies chronic liver cell injury are associated with
the development of cancer in these organs. Were it not for
the DNA repair mechanisms discussed earlier, the
incidence of chemically induced cancers in all likelihood
would be much higher. As mentioned above, the rare
hereditary disorders of DNA repair, including xeroderma
pigmentosum, are associated with greatly increased risk of
cancers induced by UV light and certain chemicals.
Radiation Carcinogenesis
Radiation, whatever its source (UV rays of sunlight, x-
rays, nuclear fission, radionuclides) is an established
carcinogen. Unprotected miners of radioactive elements
have a 10-fold increased incidence of lung cancers.
Follow-up of survivors of the atomic bombs dropped on
Hiroshima and Nagasaki disclosed a markedly increased
incidence of leukemia-principally acute and chronic
myeloid leukemia-after an average latent period of about
7 years, as well as an increased mortality rate from
thyroid, breast, colon, and lung carcinomas. The nuclear
power accident at Chernobyl in the former Soviet Union
continues to exact its toll in the form of high cancer
incidence in the surrounding areas. Therapeutic irradiation
of the head and neck can give rise to papillary thyroid
cancers years later. The oncogenic properties of ionizing
radiation are related to its mutagenic effects; it causes
chromosome breakage, translocations, and, less
frequently, point mutations. Biologically, double-stranded
DNA breaks seem to be the most important form of DNA
damage caused by radiation. There is also some evidence
that nonlethal doses of radiation may induce genomic
instability, favoring carcinogenesis.
The oncogenic effect of UV rays merits special mention
because it highlights the importance of DNA repair in
carcinogenesis. Natural UV radiation derived from the sun
can cause skin cancers (melanomas, squamous cell
carcinomas, and basal cell carcinomas). At greatest risk
are fair-skinned people who live in locales such as
Australia and New Zealand that receive a great deal of
sunlight. Nonmelanoma skin cancers are associated with
total cumulative exposure to UV radiation, whereas
melanomas are associated with intense intermittent
exposure-as occurs with sunbathing. UV light has several
biologic effects on cells. Of particular relevance to
carcinogenesis is the ability to damage DNA by forming
pyrimidine dimers. This type of DNA damage is repaired
by the nucleotide excision repair pathway. With extensive
exposure to UV light, the repair systems may be
overwhelmed, and skin cancer results. As mentioned
above, patients with the inherited disease xeroderma
pigmentosum have a defect in the nucleotide excision
repair pathway. As expected, there is a greatly increased
predisposition to skin cancers in this disorder.
Viral and Microbial Oncogenesis
a- Oncogenic RNA Viruses
The study of oncogenic retroviruses in animals has
provided spectacular insights into the genetic basis of
cancer. However, human T-cell leukemia virus-1 (HTLV-
1) is the only retrovirus that has been demonstrated to
cause cancer in humans. HTLV-1 is associated with a
form of T-cell leukemia/lymphoma that is endemic in
certain parts of Japan and the Caribbean basin but is found
sporadically elsewhere, including the United States.
Similar to the human immunodeficiency virus (HIV),
HTLV-1 has tropism for CD4+ T cells, and this subset of
T cells is the major target for neoplastic transformation.
Human infection requires transmission of infected T cells
via sexual intercourse, blood products, or breastfeeding.
Leukemia develops only in about 3% to 5% of infected
individuals after a long latent period of 20 to 50 years.
There is little doubt that HTLV-1 infection of T
lymphocytes is necessary for leukemogenesis, but the
molecular mechanisms of transformation are not clear.
HTLV-1 does not contain a viral oncogene, and in contrast
to certain animal retroviruses, no consistent integration site
next to a cellular oncogene has been discovered. Indeed, the
long latency period between initial infection and
development of disease suggests a multistep process, during
which many oncogenic mutations are accumulated.
The genome of HTLV-1 contains, in addition to the usual
retroviral genes, a unique region called pX. This region
encodes several genes, including one called TAX. The TAX

Unit 5: Neoplasia
93
protein has been shown to be necessary and sufficient for
cellular transformation. By interacting with several
transcription factors, such as NF-κB, the TAX protein can
transactivate the expression of genes that encode cytokines,
cytokine receptors, and costimulatory molecules. This
inappropriate gene expression leads to autocrine signaling
loops and increased activation of pro-mitogenic signaling
cascades. Furthermore, TAX can drive progression through
the cell cycle by directly binding to and activating cyclins.
In addition, TAX can repress the function of several tumor
suppressor genes that control the cell cycle, including
CDKN2A/p16 and p53. From these and other observations
the following scenario is emerging The TAX gene turns on
several cytokine genes and their receptors (IL-2 and IL-2R,
IL-15, and IL-15R), setting up an autocrine system that
drives T-cell proliferation. Of these cytokines, IL-15 seems
to be more important, but much remains to be defined.
Additionally, a parallel paracrine pathway is activated by
increased production of granulocyte-macrophage colony-
stimulating factor, which stimulates neighboring
macrophages to produce other T-cell mitogens. Initially the
T-cell proliferation is polyclonal because the virus infects
many cells, but, because of TAX-based inactivation of
tumor suppressor genes such as p53, the proliferating T
cells are at increased risk of secondary transforming events
(mutations), which lead ultimately to the outgrowth of a
monoclonal neoplastic T-cell population.
B - Oncogenic DNA Viruses
As with RNA viruses, several oncogenic DNA viruses
that cause tumors in animals have been identified. Four
DNA viruses-human papillomavirus (HPV), Epstein-Barr
virus (EBV), Kaposi sarcoma herpesvirus (KSHV, also
called human herpesvirus 8), and hepatitis B virus (HBV)-
are of special interest, because they are strongly
associated with human cancer
Human Papillomavirus
Scores of genetically distinct types of HPV have been
identified. Some types (e.g., 1, 2, 4, and 7) definitely cause
benign squamous papillomas (warts) in humans). By
contrast, high-risk HPVs (e.g., 16 and 18) have been
implicated in the genesis of several cancers, particularly
squamous cell carcinoma of the cervix and anogenital
region. In addition, at least 20% of oropharyngeal cancers
are associated with HPV. In contrast to cervical cancers,
genital warts have low malignant potential & are associated
with low-risk HPVs predominantly HPV-6 & HPV-11.
The oncogenic potential of HPV can be related to
products of two early viral genes, E6 and E7. Together,
they interact with a variety of growth-regulating proteins
encoded by protooncogenes and tumor suppressor genes.
The E7 protein binds to the retinoblastoma protein and
displaces the E2F transcription factors that are normally
sequestered by RB, promoting progression through the
cell cycle. Interestingly, E7 protein from high-risk HPV
types has a higher affinity for RB than does E7 from low-
risk HPV types. E7 also inactivates the CDKIs
CDKN1A/p21 and CDNK1B/p27. E7 proteins from high-
risk HPV types (types 16, 18, and 31) also bind and
presumably activate cyclins E and A. The E6 protein has
complementary effects. It binds to and mediates the
degradation of p53 and BAX, a pro-apoptotic member of
the BCL2 family, and it activates telomerase. In analogy
with E7, E6 from high-risk HPV types has a higher
affinity for p53 than E6 from low-risk HPV types.
Interestingly, in benign warts the HPV genome is
maintained in a nonintegrated episomal form, while in
cancers the HPV genome is randomly integrated into the
host genome. Integration interrupts the viral DNA,
resulting in overexpression of the oncoproteins E6 and
E7. Furthermore, cells in which the viral genome has
integrated show significantly more genomic instability.
To summarize, infection with high-risk HPV types
simulates the loss of tumor suppressor genes, activates
cyclins, inhibits apoptosis, and combats cellular
senescence. Thus, it is evident that many of the hallmarks
of cancer discussed earlier are driven by HPV proteins.
However, infection with HPV itself is not sufficient for
carcinogenesis. For example, when human keratinocytes
are transfected with DNA from HPV 16, 18, or 31 in
vitro, they are immortalized, but they do not form tumors
in experimental animals. Cotransfection with a mutated
RAS gene results in full malignant transformation. These
data strongly suggest that HPV, in all likelihood, acts in
concert with other environmental factors). However, the
primacy of HPV infection in the causation of cervical
cancer is attested to by the near complete protection from
this cancer by anti-HPV vaccines.
Epstein-Barr Virus
EBV has been implicated in the pathogenesis of several
human tumors: Burkitt lymphoma, B-cell lymphomas in
patients with acquired immunodeficiency syndrome and
other causes of immunosuppression, a subset of Hodgkin
lymphoma, and nasopharyngeal carcinoma. Except for
nasopharyngeal carcinoma, all others are B-cell tumors. A

Unit 5: Neoplasia
94
subset of T-cell lymphomas and the rare NK-cell
lymphomas may also be related to EBV.
EBV has been implicated in the pathogenesis of Burkitt
lymphomas, lymphomas in immunosuppressed
individuals with HIV infection or organ transplantation,
some forms of Hodgkin lymphoma, and nasopharyngeal
carcinoma. All except the nasopharyngeal cancers are B-
cell tumors.Certain EBV gene products contribute to
oncogenesis by stimulating a normal B-cell proliferation
pathway. Concomitant compromise of immune
competence allows sustained B-cell proliferation and
eventually development of lymphoma with occurrence of
additional mutations such as t(8 ; 14), leading to
activation of the MYC gene.
Hepatitis B and Hepatitis C Viruses:-
Between 70% and 85% of hepatocellular carcinomas
worldwide are due to infection with HBV or HCV.The
oncogenic effects of HBV and HCV are multifactorial,
but the dominant effect seems to be immunologically
mediated chronic inflammation, hepatocellular injury,
stimulation of hepatocyte proliferation, and production of
reactive oxygen species that can damage DNA.The HBx
protein of HBV and the HCV core protein can activate a
variety of signal transduction pathways that may also
contribute to carcinogenesis.
Bacterial oncogene (Helicobacter pylori)
H. pylori infection has been implicated in both gastric
adenocarcinoma and MALT lymphoma.The mechanism
of H. pylori-induced gastric cancers is multifactorial
including immunologically mediated chronic
inflammation, stimulation of gastric cell proliferation &
production of reactive oxygen species that damage DNA.
H. pylori pathogenicity genes, such as CagA, may also
contribute by stimulating growth factor pathways.It is
thought that H. pylori infection leads to polyclonal B-cell
proliferations & that eventually a monoclonal B-cell
tumor (MALT lymphoma) emerges as a result of
accumulation of mutations
Host defense against tumors: tumor
immunity
Tumor Antigens
Antigens that elicit an immune response have been
demonstrated in many experimentally induced tumors & in
some human cancers. Initially, they were broadly classified
into two categories based on their patterns of expression:
tumor-specific antigens, which are present only on tumor
cells and not on any normal cells, and tumor-associated
antigens, which are present on tumor cells and also on some
normal cells. This classification, however, is imperfect,
because many antigens thought to be tumor specific turned
out to be expressed by some normal cells as well. The
modern classification of tumor antigens is based on their
molecular structure and source. An important advance in
the field of tumor immunology was the development of
techniques for identifying tumor antigens that were
recognized by cytotoxic T lymphocytes (CTLs), because
CTLs are the major immune defense mechanism against
tumors. Recall that CTLs recognize peptides derived from
cytoplasmic proteins that are displayed bound to class I
major histocompatibility complex (MHC) molecules
Products of Other Mutated Genes
Because of the genetic instability of tumor cells, many
genes are mutated in these cells, including genes whose
products are not related to the transformed phenotype &
have no known function. Products of these mutated genes
are potential tumor antigens. These antigens are extremely
diverse because the carcinogens that induce the tumors may
randomly mutagenize virtually any host gene. Mutated
cellular proteins are found more frequently in chemical
carcinogen- or radiation-induced animal tumors than in
spontaneous human cancers. They can be targeted by the
immune system, since there is no self-tolerance against them.
Overexpressed or Aberrantly Expressed
Cellular Proteins
Tumor antigens may be normal cellular proteins that are
abnormally expressed in tumor cells and elicit immune
responses. In a subset of human melanomas some tumor
antigens are structurally normal proteins that are produced
at low levels in normal cells and overexpressed in tumor
cells. One such antigen is tyrosinase, an enzyme involved
in melanin biosynthesis that is expressed only in normal
melanocytes and melanomas. T cells from melanoma
patients recognize peptides derived from tyrosinase,

Unit 5: Neoplasia
95
raising the possibility that tyrosinase vaccines may
stimulate such responses to melanomas; clinical trials
with these vaccines are ongoing. It may be surprising that
these patients are able to respond to a normal self-antigen.
The probable explanation is that tyrosinase is normally
produced in such small amounts and in so few cells that it
is not recognized by the immune system and fails to
induce tolerance.
Another group, the so called "cancer-testis" antigens, are
encoded by genes that are silent in all adult tissues except
the testis-hence their name. Although the protein is
present in the testis it is not expressed on the cell surface
in an antigenic form, because sperm do not express MHC
class I antigens. Thus, for all practical purposes, these
antigens are tumor specific. Prototypic of this group is the
MAGE family of genes. Although they are tumor specific,
MAGE antigens are not unique for individual tumors.
MAGE-1 is expressed on 37% of melanomas and a
variable number of lung, liver, stomach, and esophageal
carcinomas. Similar antigens called GAGE, BAGE, and
RAGE have been detected in other tumors.
Tumor Antigens Produced by Oncogenic Viruses
Some viruses are associated with cancers. Not
surprisingly, these viruses produce proteins that are
recognized as foreign by the immune system. The most
potent of these antigens are proteins produced by latent
DNA viruses; examples in humans include HPV and
EBV. There is abundant evidence that CTLs recognize
antigens of these viruses and that a competent immune
system plays a role in surveillance against virus-induced
tumors because of its ability to recognize and kill virus-
infected cells. Indeed, vaccines against HPV antigens
have been found effective in prevention of cervical
cancers in young females.
Oncofetal Antigens
Oncofetal antigens or embryonic antigens, such as
carcinoembryonic antigen (CEA) and α-fetoprotein, are
expressed during embryogenesis but not in normal adult
tissues. Derepression of the genes that encode these
antigens causes their reexpression in colon and liver
cancers. Antibodies can be raised against these, and they
are useful for detection of oncofetal antigens. Although,
as discussed later, they are not entirely tumor specific,
they can serve as serum markers for cancer.
Altered Cell Surface Glycolipids & Glycoproteins
Most human and experimental tumors express higher than
normal levels and/or abnormal forms of surface
glycoproteins and glycolipids, which may be diagnostic
markers and targets for therapy. These altered molecules
include gangliosides, blood group antigens, and mucins.
Although most of the epitopes recognized by antibodies
raised against such antigens are not specifically expressed
on tumors, they are present at higher levels on cancer cells
than on normal cells. This class of antigens is a target for
cancer therapy with specific antibodies.
Several mucins are of special interest and have been the
focus of diagnostic and therapeutic studies. These include
CA-125 and CA-19-9, expressed on ovarian carcinomas,
and MUC-1, expressed on breast carcinomas. Unlike
many other types of mucins, MUC-1 is an integral
membrane protein that is normally expressed only on the
apical surface of breast ductal epithelium, a site that is
relatively sequestered from the immune system. In ductal
carcinomas of the breast, however, the molecule is
expressed in an unpolarized fashion and contains new,
tumor-specific carbohydrate and peptide epitopes. These
epitopes induce both antibody and T-cell responses in
cancer patients and are therefore being considered as
candidates for tumor vaccines.
Cell Type-Specific Differentiation Antigens
Tumors express molecules that are normally present on
the cells of origin. These antigens are called
differentiation antigens, because they are specific for
particular lineages or differentiation stages of various cell
types. Their importance is as potential targets for
immunotherapy and for identifying the tissue of origin of
tumors. For example, lymphomas may be diagnosed as B-
cell-derived tumors by the detection of surface markers
characteristic of this lineage, such as CD10 and CD20.
Antibodies against these molecules are also used for
tumor immunotherapy. These differentiation antigens are
typically normal self-antigens, and therefore they do not
induce immune responses in tumor-bearing hosts.
Antitumor Effector Mechanisms
Cell-mediated immunity is the dominant anti-tumor
mechanism in vivo. Although antibodies can be made
against tumors, there is no evidence that they play a
protective role under physiologic conditions.
Cytotoxic T Lymphocytes
The role of specifically sensitized CTLs in experimentally
induced tumors is well established. In humans, they seem to
play a protective role, chiefly against virus-associated
neoplasms (e.g., EBV-induced Burkitt lymphoma and

Unit 5: Neoplasia
96
HPV-induced tumors). The presence of MHC-restricted
CD8+ cells that can kill autologous tumor cells within
human tumors suggests that the role of T cells in immunity
against human tumors may be broader than previously
suspected. In some cases, such CD8+ T cells do not
develop spontaneously in vivo but can be generated by
immunization with tumor antigen-pulsed dendritic cells.
Natural Killer Cells
NK cells are lymphocytes that are capable of destroying
tumor cells without prior sensitization; they may provide
the first line of defense against tumor cells. After
activation with IL-2, NK cells can lyse a wide range of
human tumors, including many that seem to be
nonimmunogenic for T cells. T cells and NK cells seem to
provide complementary antitumor mechanisms. Tumors
that fail to express MHC class I antigens cannot be
recognized by T cells, but these tumors may trigger NK
cells because the latter are inhibited by recognition of
normal autologous class I molecules. The triggering
receptors on NK cells are extremely diverse and belong to
several gene families. NKG2D proteins expressed on NK
cells and some T cells are important activating receptors.
They recognize stress-induced antigens that are expressed
on tumor cells and cells that have incurred DNA damage
and are at risk for neoplastic transformation.
Macrophages
Activated macrophages exhibit cytotoxicity against tumor
cells in vitro. T cells, NK cells, and macrophages may
collaborate in antitumor reactivity, because interferon-γ, a
cytokine secreted by T cells and NK cells, is a potent
activator of macrophages. Activated macrophages may
kill tumors by mechanisms similar to those used to kill
microbes or by secretion of tumor necrosis factor (TNF).
Humoral Mechanisms
Although there is no evidence for the protective effects of
anti-tumor antibodies against spontaneous tumors,
administration of monoclonal antibodies against tumor
cells can be therapeutically effective. A monoclonal
antibody against CD20, a B cell surface antigen, is widely
used for treatment of certain non-Hodgkin lymphomas.
Immune Surveillance & Immune Evasion
by Tumors
Given the host of possible and potential antitumor
mechanisms, is there any evidence that they operate in
vivo to prevent the emergence of neoplasms? The
strongest argument for the existence of immune
surveillance is the increased frequency of cancers in
immunodeficient hosts. About 5% of individuals with
congenital immunodeficiencies develop cancers, a rate
that is about 200 times that for individuals without such
immunodeficiencies. Analogously, immunosuppressed
transplant recipients and patients with acquired
immunodeficiency syndrome have increased numbers of
malignancies. It should be noted that most (but not all) of
these neoplasms are lymphomas, often lymphomas of
activated B cells. Particularly illustrative is X-linked
lymphoproliferative disorder. When affected boys develop
an EBV infection, such infection does not take the usual
self-limited form of infectious mononucleosis but instead
evolves into a chronic or sometimes fatal form of
infectious mononucleosis or, even worse, malignant
lymphoma.
Escape mechanisms of tumor from
immunosurveillance
1) Selective outgrowth of antigen-negative variants.
During tumor progression, strongly immunogenic
subclones may be eliminated
2) Loss or reduced expression of histocompatibility
molecules.
Tumor cells may fail to express normal levels of HLA
class I, escaping attack by CTLs. Such cells, however,
may trigger NK cells
3) Immunosuppression. Many oncogenic agents (e.g.,
chemicals and ionizing radiation) suppress host immune
responses. Tumors or tumor products also may be
immunosuppressive. For example, TGF-β, secreted in
large quantities by many tumors, is a potent
immunosuppressant. In some cases, the immune response
induced by the tumor may inhibit tumor immunity.
Several mechanisms of such inhibition have been
described. For instance, recognition of tumor cells may
lead to engagement of the T-cell inhibitory receptor,
CTLA-4, or activation of regulatory T cells that suppress
immune responses.
It is worth mentioning that although much of the focus in
the field of tumor immunity has been on the mechanisms
by which the host immune system defends against tumors,
there is some recent evidence that, paradoxically, the
immune system may promote the growth of tumors. It is
possible that activated lymphocytes and macrophages
produce growth factors for tumor cells. Enzymes, such as
MMPs, that enhance tumor invasion, may also be
produced. Harnessing the protective

Unit 5: Neoplasia
97
Clinical aspects of neoplasia
Ultimately the importance of neoplasms lies in their
effects on patients. Although malignant tumors are of
course more threatening than benign tumors, any tumor,
even a benign one, may cause morbidity and mortality.
Indeed, both malignant and benign tumors may cause
problems because of (1) location and impingement on
adjacent structures, (2) functional activity such as
hormone synthesis or the development of paraneoplastic
syndromes, (3) bleeding and infections when the tumor
ulcerates through adjacent surfaces, (4) symptoms that
result from rupture or infarction & (5) cachexia or wasting
Effects of Tumor on Host
Location is crucial in both benign and malignant tumors.
A small (1-cm) pituitary adenoma can compress and
destroy the surrounding normal gland and give rise to
hypopituitarism. A 0.5-cm leiomyoma in the wall of the
renal artery may lead to renal ischemia and serious
hypertension. A comparably small carcinoma within the
common bile duct may induce fatal biliary tract obstruction.
Hormone production is seen with benign and malignant
neoplasms arising in endocrine glands. Adenomas and
carcinomas arising in the β-cells of the islets of the
pancreas can produce hyperinsulinism, sometimes fatal.
Analogously, some adenomas and carcinomas of the
adrenal cortex elaborate corticosteroids that affect the
patient (e.g., aldosterone, which induces sodium retention,
hypertension, and hypokalemia). Such hormonal activity
is more likely with a well-differentiated benign tumor
than with a corresponding carcinoma.
Cancer Cachexia
Many cancer patients suffer progressive loss of body fat
and lean body mass, accompanied by profound weakness,
anorexia, and anemia, referred to as cachexia. There is
some correlation between the size and extent of spread of
the cancer and the severity of the cachexia. However,
cachexia is not caused by the nutritional demands of the
tumor. Although patients with cancer are often anorexic,
current evidence indicates that cachexia results from the
action of soluble factors such as cytokines produced by
the tumor and the host rather than reduced food intake. In
patients with cancer, calorie expenditure remains high,
and basal metabolic rate is increased, despite reduced
food intake. This is in contrast to the lower metabolic rate
that occurs as an adaptational response in starvation. The
basis of these metabolic abnormalities is not fully
understood. It is suspected that TNF produced by
macrophages in response to tumor cells or by the tumor
cells themselves mediates cachexia. TNF suppresses
appetite and inhibits the action of lipoprotein lipase,
inhibiting the release of free fatty acids from lipoproteins.
Additionally, a protein-mobilizing factor called
proteolysis-inducing factor, which causes breakdown of
skeletal muscle proteins by the ubiquitin-proteosome
pathway, has been detected in the serum of cancer
patients. Other molecules with lipolytic action also have
been found. There is no satisfactory treatment for cancer
cachexia other than removal of the underlying cause, the
tumor.
Paraneoplastic Syndromes
Symptom complexes that occur in patients with cancer
and that cannot be readily explained by local or distant
spread of the tumor or by the elaboration of hormones
indigenous to the tissue of origin of the tumor are referred
to as paraneoplastic syndromes. They appear in 10% to
15% of patients with cancer, and it is important to
recognize them for several reasons
The paraneoplastic syndromes are diverse and are
associated with many different tumors . The most common
syndromes are hypercalcemia, Cushing syndrome, and
nonbacterial thrombotic endocarditis; the neoplasms most
often associated with these and other syndromes are lung
and breast cancers and hematologic malignancies.
Hypercalcemia in cancer patients is multifactorial, but the
most important mechanism is the synthesis of a parathyroid
hormone-related protein (PTHrP) by tumor cells. Also
implicated are other tumor-derived factors, such as TGF-α,
a polypeptide factor that activates osteoclasts, and the
active form of vitamin D. Another possible mechanism for
hypercalcemia is widespread osteolytic metastatic disease
of bone, but it should be noted that hypercalcemia resulting
from skeletal metastases is not a paraneoplastic syndrome.
Cushing syndrome as a paraneoplastic phenomenon is
usually related to ectopic production of ACTH or ACTH-
like polypeptides by cancer cells, as occurs in small-cell
cancers of the lung. Sometimes one tumor induces several
syndromes concurrently. For example, bronchogenic
carcinomas may elaborate products identical to or having
the effects of ACTH, antidiuretic hormone, parathyroid
hormone, serotonin, human chorionic gonadotropin, and
other bioactive substances.

Unit 5: Neoplasia
98
Grading and Staging of Cancer:-
Methods to quantify the probable clinical aggressiveness of
a given neoplasm and its apparent extent and spread in the
individual patient are necessary for making accurate
prognosis and for comparing end results of various
treatment protocols. For instance, the results of treating
extremely small, highly differentiated thyroid
adenocarcinomas that are localized to the thyroid gland are
likely to be different from those obtained from treating
highly anaplastic thyroid cancers that have invaded the
neck organs.
The grading of a cancer attempts to establish some
estimate of its aggressiveness or level of malignancy
based on the cytologic differentiation of tumor cells and
the number of mitoses within the tumor. The cancer may
be classified as grade I, II, III, or IV, in order of
increasing anaplasia. Criteria for the individual grades
vary with each form of neoplasia and so are not detailed
here. Difficulties in establishing clear-cut criteria have led
in some instances to descriptive characterizations (e.g.,
"well-differentiated adenocarcinoma with no evidence of
vascular or lymphatic invasion" or "highly anaplastic
sarcoma with extensive vascular invasion").
Staging of cancers is based on the size of the primary
lesion, its extent of spread to regional lymph nodes, and
the presence or absence of metastases. This assessment is
usually based on clinical and radiographic examination
(computed tomography and magnetic resonance imaging)
and in some cases surgical exploration. Two methods of
staging are currently in use: the TNM system (T, primary
tumor; N, regional lymph node involvement; M,
metastases) and the AJC (American Joint Committee)
system. In the TNM system, T1, T2, T3, and T4 describe
the increasing size of the primary lesion; N0, N1, N2, and
N3 indicate progressively advancing node involvement;
and M0 and M1 reflect the absence or presence of distant
metastases. In the AJC method, the cancers are divided
into stages 0 to IV, incorporating the size of primary
lesions and the presence of nodal spread and of distant
metastases. Examples of the application of these two
staging systems are cited in subsequent chapters. It is
worth noting that when compared with grading, staging
has proved to be of greater clinical value.

Unit 5: Neoplasia
99
Laboratory Diagnosis of Cancer

Unit 5: Neoplasia
100