
48

Unit 4: Diseases of the Immune System
49
Innate and adaptive immunity
Defense against microbes consists of 2 types of reactions.
Innate immunity (also called natural, or native, immunity)
is mediated by cells and proteins that are always present
& fight against microbes & action immediately in
response to infection
The major components of innate immunity are:
epithelial barriers of the skin,
gastrointestinal tract,
respiratory tract, which prevent microbe entry
phagocytic leukocytes (neutrophils and macrophages);
natural killer (NK) cell;
Several circulating plasma proteins, the most important
of which are the proteins of the complement system.
However, many pathogenic microbes have evolved to
overcome innate immune defenses, and protection against
these infections requires the more powerful mechanisms
of adaptive immunity (also called acquired, or specific,
immunity).
Adaptive immunity is normally silent and "adapts” to the
presence of infectious microbes by becoming active,
expanding, and generating potent mechanisms for
neutralizing and eliminating the microbes.
The components of the adaptive immune system are
lymphocytes and their products.
There are two types of adaptive immune responses:
humoral immunity, mediated by soluble antibody proteins
that are produced by B lymphocytes (also called B cells
cell-mediated (or cellular) immunity, mediated by T
lymphocytes (also called T cells)
Antibodies provide protection against extracellular
microbes in the blood, mucosal secretions, and tissues.
T lymphocytes are important in defense against
intracellular microbes.
They work by either directly killing infected cells
(accomplished by cytotoxic T lymphocytes) or by
activating phagocytes to kill ingested microbes, via the
production of soluble protein mediators called cytokines
(made by helper T cells).
When the immune system is inappropriately triggered or
not properly controlled, the same mechanisms that are
involved in host defense cause tissue injury and disease.
The reaction of the cells of innate and adaptive immunity
may be manifested as inflammation.
Inflammation is a beneficial process, but it is also the
basis of many human diseases.
Cells & Tissues of the Immune System
Lymphocytes
Lymphocytes: are present in the circulation and in various
lymphoid organs.
T lymphocytes are so called because they mature in the
thymus, whereas B lymphocytes mature in the bone
marrow.
They are recognizing tens or hundreds of millions of
antigens. This enormous diversity of antigen recognition
is generated by the somatic rearrangement of antigen
receptor genes during lymphocyte maturation, and
variations that are introduced during the joining of
different gene segments to form antigen receptors
Unit 4: Diseases of the Immune System
50
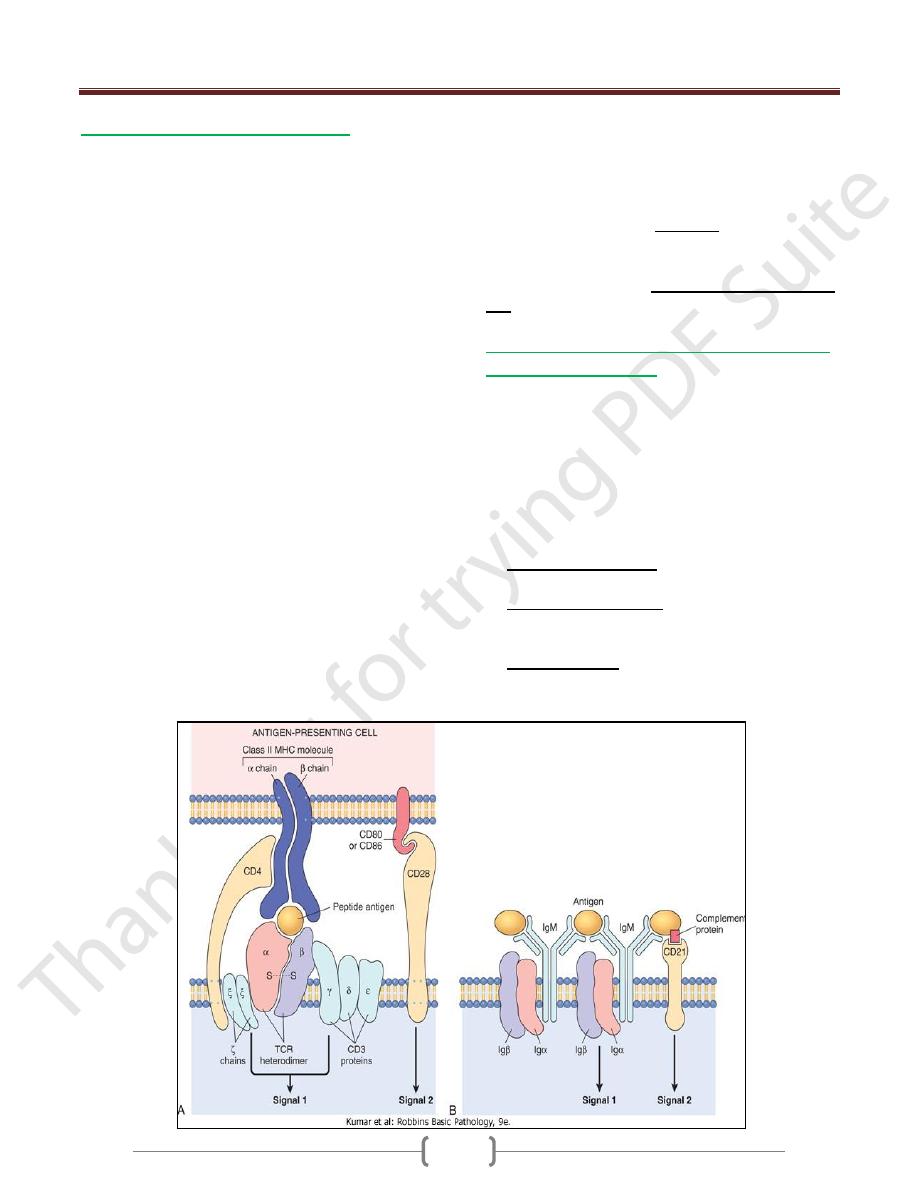
1- T (thymus-derived) lymphocytes
Express Ag receptors called T cell receptors (TCRs) that
recognize peptide fragments of protein antigens that are
displayed by MHC molecules on the surface of antigen-
presenting cells.
T cells recognize only peptide fragments of protein
antigens that are displayed on other cells bound to
proteins of the major histocompatibility complex (MHC;
or in humans, human leukocyte antigen [HLA] complex
the normal function of MHC molecules is to display
peptides for recognition by T lymphocytes.
In every individual, T cells recognize only peptides
displayed by that individual's MHC molecules, which, of
course, are the only MHC molecules that the T cells will
encounter normally. This phenomenon is called MHC
restriction.
TCRs are noncovalently linked to a cluster of five
invariant polypeptide chains, the γ, δ, and ε proteins of the
CD3 molecular complex and two ζ chains
T cells express a number of other invariant function-
associated molecules. CD4 & CD8 are expressed on distinct
T-cell subsets & serve as coreceptors for T-cell activation
CD4+ T cells are "helper" T cells because they secrete
soluble molecules (cytokines) that help B cells to produce
antibodies (the origin of the name "helper" cells) and also
help macrophages to destroy phagocytosed microbes.
CD8+ T cells can also secrete cytokines, but they play a
more important role in directly killing virus-infected or
tumor cells, and hence are called "cytotoxic" T
lymphocytes (CTLs). Other important invariant proteins
on T cells include CD28, which functions as the receptor
for molecules that are induced on APCs by microbes (and
are called costimulators),
Another small population of T cells expresses markers of
T cells and NK cells. These NKT cells recognize
microbial glycolipids
Another populations of T cells that functions to suppress
immune response is that of regulatory T lymphocytes (T
reg)
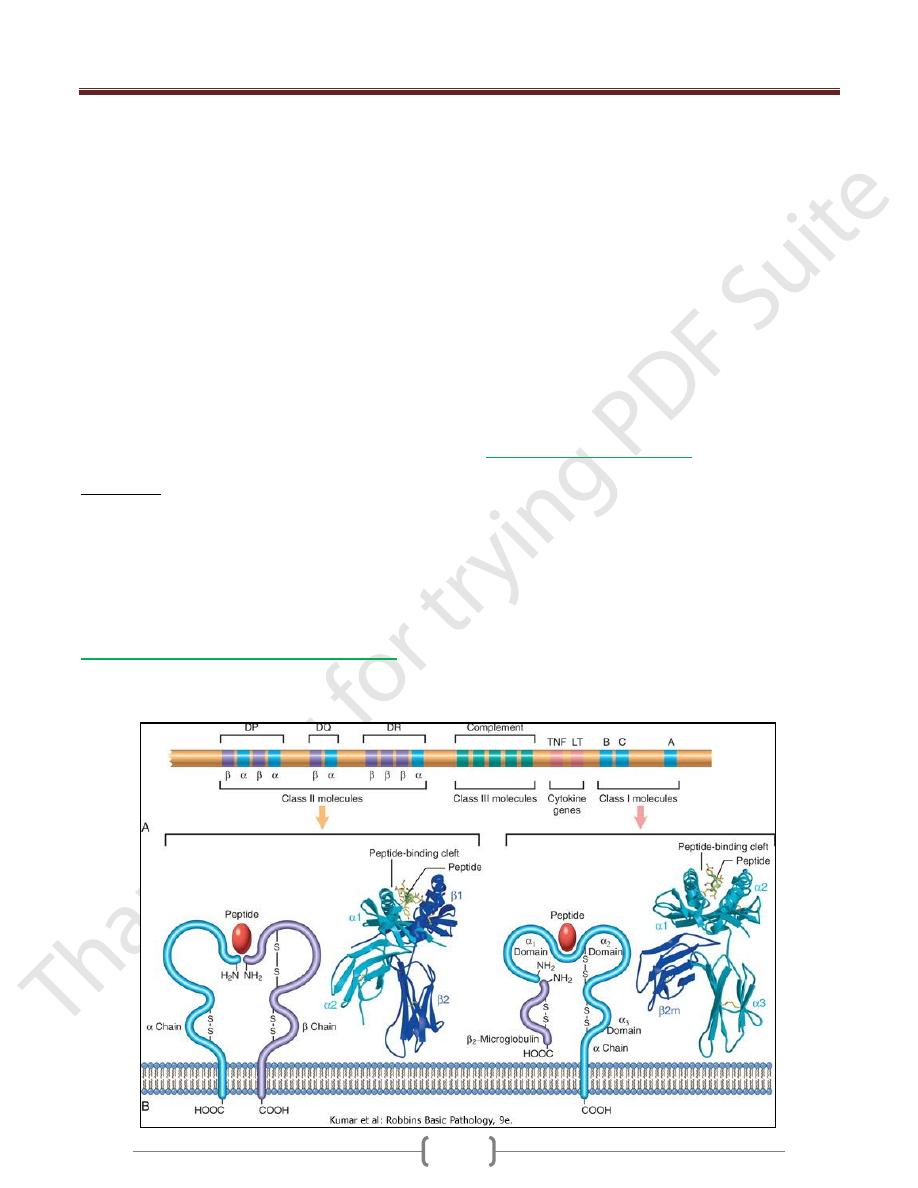
MHC Molecules: the Peptide Display System
of Adaptive Immunity
The human MHC, known as the human leukocyte antigen
(HLA) complex, consists of a cluster of genes on chrom. 6
The HLA system is highly polymorphic
this polymorphism also constitutes a formidable barrier to
organ transplantation
On the basis of their chemical structure, tissue
distribution, and function, MHC gene products fall into
three categories:
Class I MHC molecules are encoded by three closely
linked loci, designated HLA-A, HLA-B, and HLA-C
Class II MHC molecules are encoded by genes in the
HLA-D region, which contains at least three
subregions: DP, DQ, and DR.
Class III proteins include some of the complement
components (C2, C3, and Bf); genes encoding tumor
necrosis factor (TNF) and lymphotoxin (LT)
Unit 4: Diseases of the Immune System
51
Different MHC alleles bind to different peptide fragments
depending on the particular amino acid sequence of a
given peptide; the expression of many different MHC
molecules allows each cell to present a wide array of
peptide antigens.
The role of the MHC in T-cell stimulation also has
important implications for the genetic control of immune
responses.
The ability of any given MHC allele to bind the peptide
antigens generated from a particular pathogen will
determine whether an individual's T cells can actually
"see" and respond to that pathogen.
An individual will recognize and mount an immune
response against a given antigen only if he or she inherits
MHC molecules that can bind the antigenic peptide and
present it to T cells.
The inheritance of particular alleles influences both
protective and harmful immune responses.
For example, if the antigen is pollen and the response is
an allergic reaction,
Inheritance of some HLA genes may make individuals
susceptible to this disease.
On the other hand, good responsiveness to a viral antigen,
determined by inheritance of certain HLA alleles, may be
beneficial for the host.
2- B (bone marrow-derived) lymphocytes
Express membrane-bound antibodies that recognize a
wide variety of antigens.
B cells are activated to become plasma cells, which
secrete antibodies.
They are also present in bone marrow and in the follicles
of peripheral lymphoid tissues (lymph nodes, spleen,
tonsils, and other mucosal tissues).
B cells express several invariant molecules that are
responsible for signal transduction & for activation of the
cells.
These molecules include the CD40 receptor, which binds
to its ligand expressed on helper T cells, and CD21 (also
known as the CR2 complement receptor), which
recognizes a complement breakdown product that is
frequently deposited on microbes
EBV use CD21 as a receptor for the binding to B cell and
infecting them
3- Natural killer (NK) cells
Kill cells that are infected by some microbes.
It has 2 types of receptors: activating & inhibitory receptors
To avoid attacking normal host cells, NK cells express
inhibitory receptors that recognize self class I MHC
molecules, which are expressed on all healthy cells;
engagement of these inhibitory receptors typically
overrides the activating receptors and thus prevents
activation of the NK cells.
NK cells express inhibitory receptors that recognize MHC
molecules that are normally expressed on healthy cells,
and are thus prevented from killing normal cells.
Unit 4: Diseases of the Immune System
52
Antigen-presenting cells (APCs)
Capture microbes and other antigens, transport them to
lymphoid organs, and display them for recognition by
lymphocytes.
The most efficient APCs are:
Dendritic cells,
Cells with dendritic morphology (i.e., with fine dendritic
cytoplasmic processes) occur as two functionally distinct
types.
Interdigitating DCs, or more simply, DCs, are
nonphagocytic cells that express high levels of class II
MHC and T-cell costimulatory molecules
It is live in and under epithelia in most tissues.
1) One subset of DC is called Plasmacytoid DCs because of
their resemblance to plasma cells
They present in the blood and lymphoid organs
They are major source of antiviral cytokines type I
interferon produce to many viruses
2) The second type of DCs is called follicular dendritic
cells (FDCs). They are located in the germinal centers of
lymphoid follicles in the spleen and lymph nodes. These
cells bear receptors for the Fc tails of IgG and for
complement proteins, and hence efficiently trap antigen
bound to antibodies and complement. These cells display
antigens to activated B lymphocytes in lymphoid follicles
and promote secondary antibody responses.
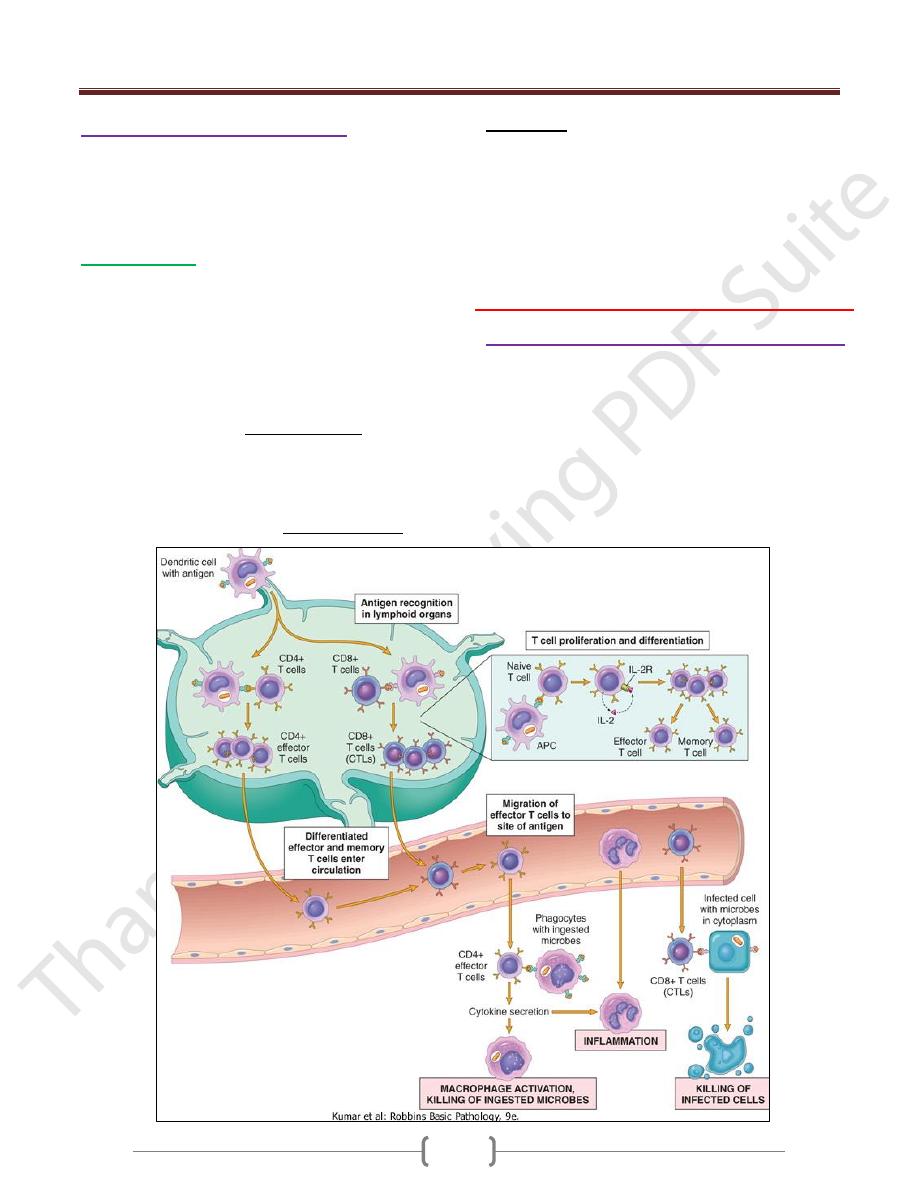
Overview of normal immune responses
The early innate immune response to microbes
The physiologic function of the immune system is defense
against infectious microbes. The early reaction to microbes
is mediated by the mechanisms of innate immunity, which
are ready to respond to microbes. These mechanisms
include epithelial barriers, phagocytes, NK cells, and
plasma proteins, e.g., of the complement system.
The reaction of innate immunity is often manifested as
inflammation.

Unit 4: Diseases of the Immune System
53
The Capture & Display of Microbial Antigens
The defense reactions of adaptive immunity develop
slowly, but are more potent and specialized.
Microbes and other foreign antigens are captured by
dendritic cells and transported to lymph nodes, where the
antigens are recognized by naïve lymphocytes.
The lymphocytes are activated to proliferate and
differentiate into effector and memory cells.
Cell-Mediated Immunity: Activation of T
Lymphocytes & Elimination of Cell-
Associated Microbes
Cell-mediated immunity is the reaction of T lymphocytes,
designed to combat cell-associated microbes (e.g.,
phagocytized microbes and microbes in the cytoplasm of
infected cells).
Humoral immunity is mediated by antibodies and is
effective against extracellular microbes (in the circulation
and mucosal lumens).
Cells neutralize microbes and block their infectivity, and
promote the Antibodies secreted by plasma phagocytosis
and destruction of pathogens. Antibodies also confer
passive immunity to neonates.
CD4+ helper T cells help B cells to make antibodies,
activate macrophages to destroy ingested microbes, and
regulate all immune responses to protein antigens. The
functions of CD4+ T cells are mediated by secreted
proteins called cytokines.
CD8+ cytotoxic T lymphocytes kill cells that express
antigens in the cytoplasm that are seen as foreign (e.g.
virus-infected and tumor cells).
Cytokines
Are Messenger Molecules of the Immune System Body”
Cytokines are polypeptide products of many cell types
(but principally activated lymphocytes and macrophages)
that function as mediators of inflammation and immune
responses
Although different cytokines have diverse actions and
functions, they all share some common features.
Cytokines are synthesized and secreted in response to
external stimuli, which may be microbial products,
antigen recognition, and other cytokines.
Their secretion is typically transient and is controlled by
transcription and post-translational mechanisms.
The actions of cytokines may be
autocrine (on the cell that produces the cytokine),
paracrine (on adjacent cells), and, less commonly,
Endocrine (at a distance from the site of production).
The effects of cytokines tend to be
Pleiotropic (one cytokine has effects on many cell types)
Redundant (the same activity may be induced by many
proteins).
Cytokines may be grouped into several classes based
on their biologic activities and functions.
1) Cytokines involved in innate immunity and
inflammation (IL-1,IL-12,IL-6. IL-23.IFN TNF)
2) Cytokines that regulate lymphocyte responses and
effector functions in adaptive immunity(IL-2, IL-4)
3) Cytokines that stimulate hematopoiesis. Many of these
are called colony-stimulating factors
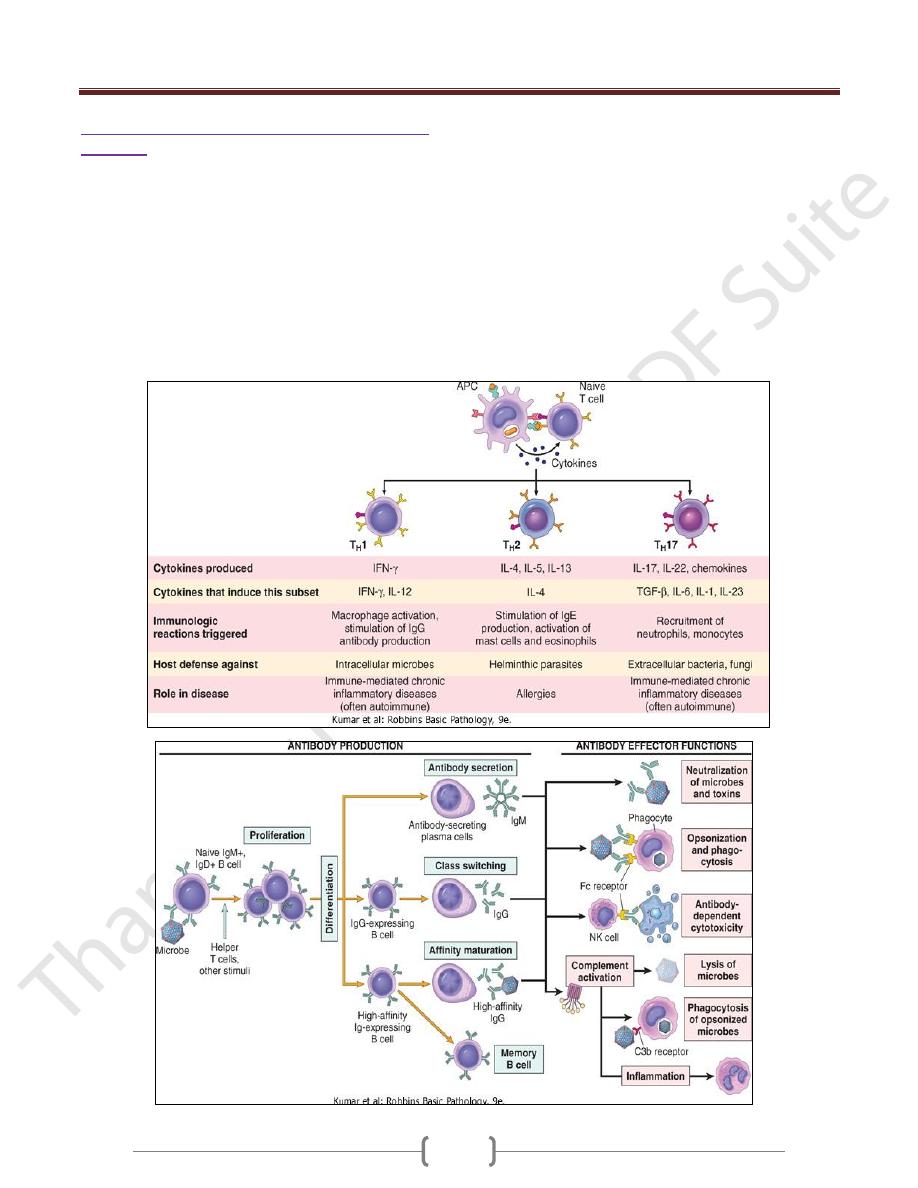
Effector Functions of T Lymphocytes
The best defined subsets of CD4+ helper cells are the T
H
1
and T
H
2 and TH17subsets
One of the earliest responses of CD4+ helper T cells is
secretion of the cytokine IL-2 and expression of high-
affinity receptors for IL-2. IL-2 is a growth factor that acts
on these T lymphocytes and stimulates their proliferation,
leading to an increase in the number of antigen-specific
lymphocytes.
A third subset, called T
H
17, has been described recently
that produces the cytokine IL-17, which promotes
inflammation, and is believed to play an important role in
some T cell-mediated inflammatory disorders.
These effector cells migrate to sites of infection and
accompanying tissue damage.
When the differentiated effectors again encounter cell-
associated microbes, they are activated to perform the
functions that are responsible for elimination of the
microbes.
The key mediators of the functions of helper T cells are
the surface molecule called CD40 ligand (CD40L), which
binds to its receptor, CD40, on B cells and macrophages,
Humoral immunity: activation of B lymphocytes
& elimination of extracellular microbes
Upon activation, B lymphocytes proliferate and then
differentiate into plasma cells that secrete different classes
of antibodies with distinct functions
B cells can also act as APCs, i.e. ingest protein antigens,
degrade them, and display peptides bound to class II
MHC molecules for recognition by helper T cells. The
helper T cells express CD40L and secrete cytokines,
which work together to activate the B cells.
Unit 4: Diseases of the Immune System
54
Decline of immune responses & Immunologic
Memory
The majority of effector lymphocytes induced by an
infectious pathogen die by apoptosis after the microbe is
eliminated, thus returning the immune system to its basal
resting state.
This return to a stable or steady state is called homeostasis.
It occurs because microbes provide essential stimuli for
lymphocyte survival and activation and effector cells are
short-lived.
Therefore, as the stimuli are eliminated, the activated
lymphocytes are no longer kept alive
The initial activation of lymphocytes also generates long-
lived memory cells, which may survive for years after the
infection.
Memory cells are an expanded pool of antigen-specific
lymphocytes (more numerous than the naive cells specific
for any antigen that are present before encounter with that
antigen), and memory cells respond faster and more
effectively against the antigen than do naive cells. This is
why the generation of memory cells is an important goal
of vaccination.

Unit 4: Diseases of the Immune System
55
Hypersensitivity reactions:
mechanisms of immune-mediated
injury
Immune responses are capable of causing tissue injury
and diseases that are called hypersensitivity diseases.
This term originated from the idea that individuals who
mount immune responses against an antigen are said to be
"sensitized" to that antigen, and therefore, pathologic or
excessive reactions are manifestations of "hypersensitivity
Normally, an immune system of checks and eradicate the
infecting organisms without serious injury to host tissues.
However, immune responses may be inadequately
controlled or inappropriately targeted to host tissues, and
in these situations, the normally beneficial response is the
cause of disease.
Causes of Hypersensitivity Diseases
Pathologic immune responses may be directed against
different types of antigens, and may result from various
underlying abnormalities:
1) Autoimmunity
Normally, the immune system does not react against an
individual's own antigens. This phenomenon is called self-
tolerance, implying that all of us "tolerate" our own
antigens. Sometimes, self-tolerance fails, resulting in
reactions against one's own cells and tissues that are
called autoimmunity.
2) Reactions against microbial antigens
that may cause disease. In some cases, the reaction
appears to be excessive or the microbial antigen is
unusually persistent.
If antibodies are produced against such antigens, the
antibodies may bind to the microbial antigens to produce
immune complexes, which deposit in tissues and trigger
inflammation; this is the underlying mechanism of
poststreptococcal glomerulonephritis
T-cell responses against persistent microbes may give rise
to severe inflammation, sometimes with the formation of
granulomas this is the cause of tissue injury in
tuberculosis and other infections.
Rarely, antibodies or T cells reactive with a microbe
cross-react with a host tissue; this is believed to be the
basis of rheumatic heart disease
3) Reactions against environmental antigens.
Most healthy individuals do not react strongly against
common environmental substances (e.g., pollens, animal
danders, or dust mites), but almost 20% of the population is
"allergic" to these substances
Allergies are diseases caused by unusual immune responses
to a variety of noninfectious,
Types of Hypersensitivity Diseases
Hypersensitivity reactions are traditionally subdivided
into four types; three are variations on antibody-mediated
injury, whereas the fourth is cell mediated
Immediate (Type I) Hypersensitivity
Often called allergy,
Results from the activation of the T
H
2 subset of CD4+
helper T cells by environmental antigens, leading to the
production of IgE antibodies, which become attached to
mast cells.
When these IgE molecules bind the antigen (allergen), the
mast cells are triggered to release mediators that transiently
affect vascular permeability & induce smooth muscle
contraction in various organs & that also may stimulate more
prolonged inflammation (the late-phase reaction). These
diseases are commonly called allergic, or atopic, disorders.
This type is a tissue reaction that occurs rapidly (typically
within minutes) after the interaction of antigen with IgE
antibody that is bound to the surface of mast cells in a
sensitized host.
The reaction is initiated by entry of an antigen, which is
called an allergen because it triggers allergy.
Many allergens are environmental substances that are
harmless for most persons on exposure.
Some people apparently inherit genes that make them
susceptible to allergies.
This susceptibility is manifested by the ability of such
persons to mount strong T
H
2 responses and, subsequently,
to produce IgE antibody against the allergens.
The IgE is central to the activation of the mast cells and
release of mediators that are responsible for the clinical
and pathologic manifestations of the reaction.
Immediate hypersensitivity may occur as a local reaction
that is merely annoying (e.g., seasonal rhinitis or hay fever),
severely debilitating (asthma), or even fatal (anaphylaxis).
Sequence of Events in Immediate Hypersensitivity
Reactions
Most hypersensitivity reactions follow the same
sequence of cellular responses:
Activation of T
H
2 cells and production of IgE antibody.
Allergens may be introduced by inhalation, ingestion,
or injection.

Unit 4: Diseases of the Immune System
56
Variables that probably contribute to the strong T
H
2
responses to allergens include:
The route of entry,
dose,
chronicity of antigen exposure,
The genetic makeup of the host.
Structural properties that endow them with the ability
to elicit T
H
2 responses.
The TH2 cells that are induced secrete several cytokines:
IL-4 stimulates B cells specific for the allergen to undergo
heavy-chain class switching to IgE and to secrete this
immunoglobulin isotype.
IL-5 activates eosinophils that are recruited to the reaction,
IL-13 acts on epithelial cells & stimulates mucus secretion.
TH2 cells often are recruited to the site of allergic reactions
in response to chemokines that are produced locally
Chemokines is eotaxin, which also recruits eosinophils to
the same site.
Sensitization of mast cells by IgE antibody.
Mast cells are derived from precursors in the bone marrow,
are widely distributed in tissues, and often reside near blood
vessels and nerves and in subepithelial locations.
Mast cells express a high-affinity receptor for the Fc
portion of the Ε heavy chain of IgE, called FcΕRI.
These antibody-bearing mast cells are "sensitized" to react
if the antigen binds to the antibody molecules.
Basophiles
They also express FcΕRI, but their role in most immediate
hypersensitivity reactions is not established (since these
reactions occur in tissues and not in the circulation).
Eosinophils
The third cell type that expresses FcΕRI , which often are
present in these reactions and also have a role in IgE-
mediated host defense against helminth infections,
Activation of mast cells and release of mediators. When a
person who was sensitized by exposure to an allergen is
reexposed to the allergen, it binds to multiple specific IgE
molecules on mast cells, usually at or near the site of
allergen entry.
When these IgE molecules are cross-linked, a series of
biochemical signals is triggered in the mast cells. The
signals culminate in the secretion of various mediators
from the mast cells.
3 groups of mediators are the most important in
different immediate hypersensitivity reactions:
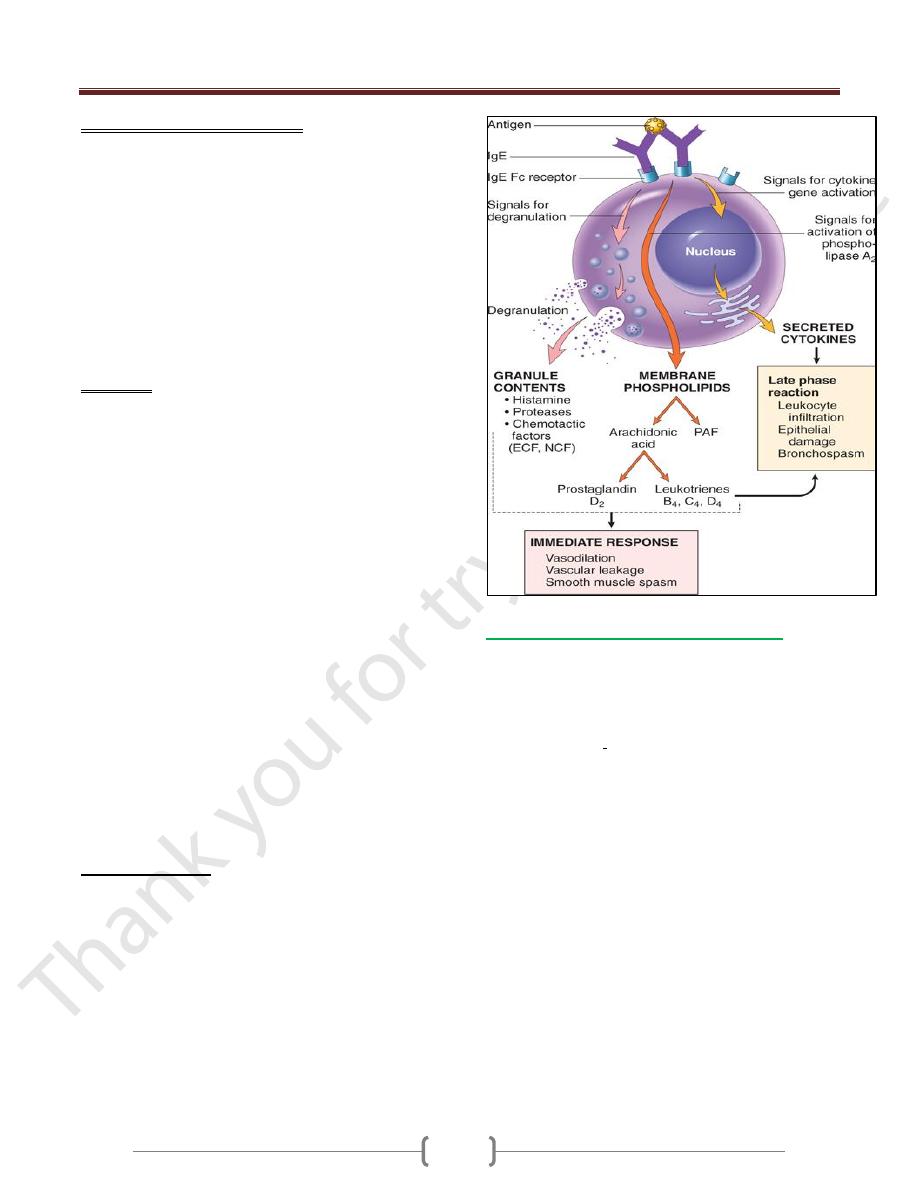
1) Vasoactive amines released from granule stores.
The granules of mast cells contain
a) Histamine, which is released within seconds or
minutes of activation. Histamine causes vasodilation,
increased vascular permeability, smooth muscle
contraction, and increased secretion of mucus.
b) 2-neutral proteases (e.g., tryptase), which may damage
tissues and also generate kinins and cleave
complement components to produce additional
chemotactic and inflammatory factors (e.g., C3a)
Unit 4: Diseases of the Immune System
57
2) Newly synthesized lipid mediators.
Mast cells synthesize and secrete prostaglandins and
leukotrienes,
These lipid mediators have several actions that are
important in immediate hypersensitivity reactions.
a) Prostaglandin D
2
(PGD
2
) is the most abundant mediator
generated by the cyclooxygenase pathway in mast cells. It
causes intense bronchospasm as well as increased mucus
secretion.
b) The leukotrienes LTC
4
and LTD
4
are the most potent
vasoactive and spasmogenic agents known; LTB
4
is
highly chemotactic for neutrophils, eosinophils, and
monocytes.
3) Cytokines.
Activation of mast cells results in the synthesis and
secretion of several cytokines that are important for the
late-phase reaction. These include TNF and chemokines,
which recruit and activate leukocytes
IL-4 and IL-5, which amplify the T
H
2-initiated immune
reaction; and IL-13, which stimulates epithelial cell
mucus secretion.
Often, the IgE-triggered reaction has 2 well-defined phases:
a) The immediate response, characterized by vasodilation,
vascular leakage, and smooth muscle spasm, usually
evident within 5 to 30 minutes after exposure to an
allergen and subsiding by 60 minutes;
b) a second, late-phase reaction
that usually sets in 2 to 8 hours later and may last for
several days
Characterized by inflammation as well as tissue
destruction, such as mucosal epithelial cell damage.
The dominant inflammatory cells in the late-phase
reaction are neutrophils, eosinophils, and lymphocytes,
especially T
H
2 cells.
Eosinophils are recruited by eotaxin and other
chemokines
Eosinophils produce
1) major basic protein
2) Eosinophil cationic protein, which are toxic to epithelial
cells
3) LTC
4
4) Platelet-activating factor, which promote inflammation.
T
H
2 cells produce cytokines
Because inflammation is a major component of many
allergic diseases, notably asthma and atopic dermatitis,
therapy usually includes anti-inflammatory drugs such as
corticosteroids.
Clinical and Pathologic Manifestations
An immediate hypersensitivity reaction occurs as a
systemic disorder or as a local reaction.
The nature of the reaction is often determined by the route
of antigen exposure.
Systemic exposure to protein antigens (e.g., in bee
venom) or drugs (e.g., penicillin) may result in systemic
anaphylaxis. urticaria (hives), and skin erythema appear,
followed in short order by profound respiratory difficulty
caused by pulmonary bronchoconstriction and
accentuated by hypersecretion of mucus. the musculature
of the entire gastrointestinal tract may be affected, with
resultant vomiting, abdominal cramps, and diarrhea.
(Anaphylactic shock), and the patient may progress to
circulatory collapse and death within minutes.
Local reactions generally occur when the antigen is
confined to a particular site, such as:
Skin (contact, causing urticaria),
Gastrointestinal tract (ingestion, causing diarrhea),
Lung (inhalation, causing bronchoconstriction).
Nose, (inhalation)hay fever,
Patients who suffer from allergy often have a family
history of similar conditions.
Genes involved in:

Unit 4: Diseases of the Immune System
58
encoding HLA molecules
cytokines (which may control T
H
2 responses),
a component of the FcΕRI,
The immune response dependent on T
H
2 cells and IgE-in
particular, the late-phase inflammatory reaction-plays an
important protective role in combating parasitic infections.
IgE antibodies are produced in response to many
helminthic infections, and their physiologic function is to
target helminths for destruction by eosinophils & mast cells
Antibody-Mediated Diseases (Type II
Hypersensitivity)
Antibody-mediated (type II) hypersensitivity disorders are
caused by
Antibodies that bind to fixed tissue or cell surface antigens,
promoting phagocytosis and destruction of the coated cells
or triggering pathologic inflammation in tissues.
Antibodies directed against: target antigens on the surface
of cells or other tissue components. The antigens may be
normal molecules intrinsic to cell membranes or in the
extracellular matrix, they may be adsorbed exogenous
antigens (e.g., a drug metabolite).
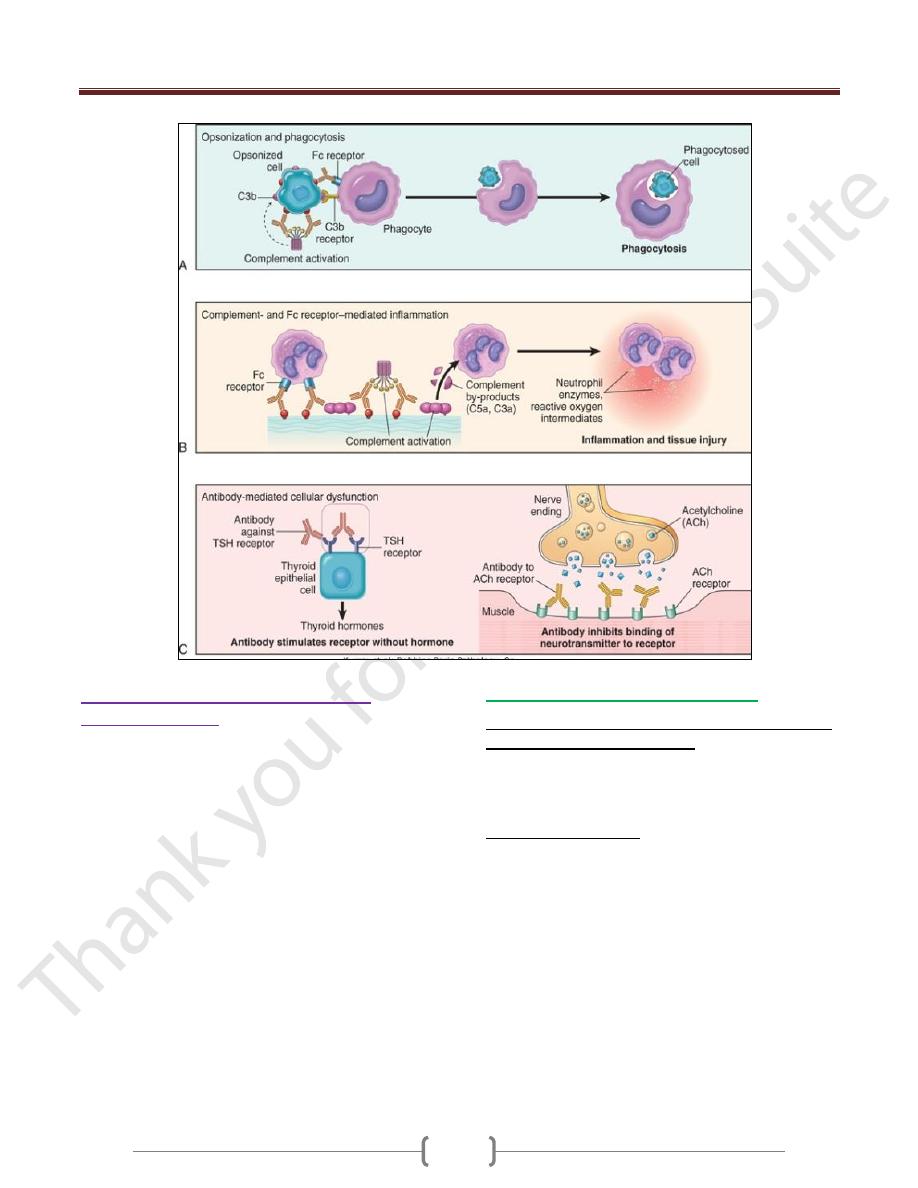
Mechanisms of Antibody-Mediated Diseases
Antibodies cause disease by targeting cells for
phagocytosis, and by interfering with normal cellular
functions
The antibodies that are responsible typically are high-
affinity antibodies capable of activating complement and
binding to the Fc receptors of phagocytes.
1) Opsonization and phagocytosis.
When circulating cells, such as erythrocytes or platelets,
are coated (opsonized) with autoantibodies, with or
without complement proteins, the cells become targets for
phagocytosis by neutrophils and macrophages
These phagocytes express receptors for the Fc tails of IgG
antibodies and for breakdown products of the C3
complement protein, and use these receptors to bind and
ingest opsonized particles.
Opsonized cells are usually eliminated in the spleen, and
this is why splenectomy is of clinical benefit in
autoimmune thrombocytopenia and some forms of
autoimmune hemolytic anemia
2)
Antibodies bound to cellular or tissue antigens
activate the complement system by the "classical"
pathway end with inflammation
Products of complement activation serve several functions
of which is to recruit neutrophils and monocytes,
triggering inflammation in tissues.
3) Antibody-mediated cellular dysfunction.
In some cases, antibodies directed against cell surface
receptors impair or dysregulate cellular function without
causing cell injury or inflammation.
In myasthenia gravis, antibodies against acetylcholine
receptors in the motor end plates of skeletal muscles inhibit
neuromuscular transmission, with resultant muscle weakness.
In Graves disease, Abs against the thyroid-stimulating
hormone receptor stimulate thyroid epithelial cells to
secrete thyroid hormones, resulting in hyperthyroidism.
Antibodies against hormones and other essential proteins
can neutralize and block the actions of these molecules,
causing functional derangements
Unit 4: Diseases of the Immune System
59
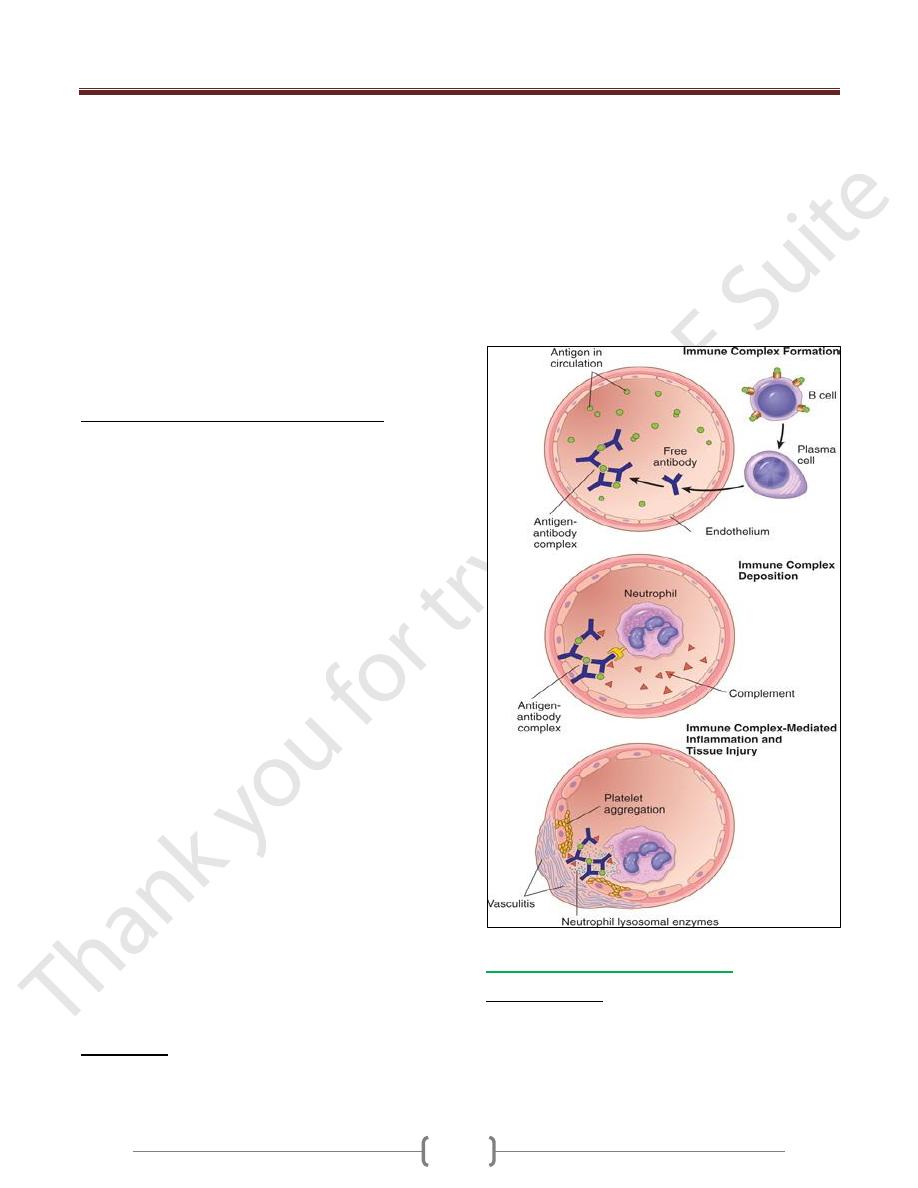
Immune complex diseases (type III
hypersensitivity)
Are caused by antibodies binding to antigens to form
immune complexes that circulate and deposit in vascular
beds and stimulate inflammation, typically as a
consequence of complement activation. Tissue injury in
these diseases is the result of the inflammation.
Antigen-antibody (immune) complexes that are formed in
the circulation may deposit in blood vessels, leading to
complement activation and acute inflammation.
The antigens in these complexes may be exogenous
antigens, such as microbial proteins, or endogenous
antigens, such as nucleoproteins.
Small amounts of antigen-antibody complexes may be
produced during normal immune responses and are
usually phagocytosed and destroyed.
Immune complex-mediated injury is systemic when
complexes are formed in the circulation and are deposited
in several organs, or it may be localized to particular
organs (e.g., kidneys, joints, or skin) if the complexes are
formed and deposited in a specific site.
Systemic Immune Complex Disease
The pathogenesis of systemic immune complex disease
can be divided into three phases:
(1) formation of antigen-antibody complexes in the circulation
(2) deposition of the immune complexes in various tissues
(3) inflammatory reaction in various sites throughout the body
Acute serum sickness
It was first described in humans when large amounts of
foreign serum were administered for passive immunization
(e.g., in persons receiving horse serum containing
antidiphtheria antibody
Approximately 5 days after the foreign protein is injected,
specific antibodies are produced; these react with the
antigen still present in the circulation to form antigen-
antibody complexes.
The complexes deposit in blood vessels in various tissue
beds, triggering the subsequent injurious inflammatory
reaction.
Several variables determine whether immune complex
formation leads to tissue deposition and disease.
Unit 4: Diseases of the Immune System
60
1) The size of the complexes. Very large complexes or
complexes with many free IgG Fc regions (typically
formed in antibody excess) are rapidly removed from the
circulation by macrophages in the spleen and liver and are
therefore usually harmless.
2) The charge of the complex,
3) The valency of the antigen,
4) The avidity of the antibody, 5-the hemodynamics of a
given vascular bed
The favored sites of deposition are:
Kidneys, joints, and small blood vessels in many tissues.
Localization in the kidney and joints is explained in part
by the high hemodynamic pressures associated with the
filtration function of the glomerulus and the synovium.
The third phase, the inflammatory reaction.
During this phase (approximately 10 days after antigen
administration), clinical features such as fever, urticaria,
arthralgias, lymph node enlargement, and proteinuria
appear.
The immune complexes activate the complement system,
leading to the release of biologically active fragments
such as the anaphylatoxins (C3a and C5a), which increase
vascular permeability and are chemotactic for neutrophils
and monocytes
The complexes also bind to Fcγ receptors on neutrophils
and monocytes, activating these cells.
phagocytosis of immune complexes by the leukocytes
results in the secretion of a variety of additional pro-
inflammatory substances, including prostaglandins,
vasodilator peptides, and chemotactic substances, as well
as lysosomal enzymes capable of digesting basement
membrane, collagen, elastin, and cartilage, and reactive-
oxygen species that damage tissues.
Immune complexes can also cause platelet aggregation
and activate Hageman factor; both of these reactions
augment the inflammatory process and initiate formation
of microthrombi, which contribute to the tissue injury by
producing local ischemia
The resultant pathologic lesion is termed vasculitis if it
occurs in blood vessels, glomerulonephritis if it occurs in
renal glomeruli,arthritis if it occurs in the joints, & so on.
(IgG and IgM) and antibodies that bind to phagocyte Fc
receptors (IgG). During the active phase of the disease,
consumption of complement may result in decreased
serum complement levels.
Morphology
:
The morphologic appearance of immune complex injury
is dominated by acute necrotizing
vasculitis, microthrombi, and superimposed ischemic
necrosis accompanied by acute inflammation of the
affected organs.
The necrotic vessel wall takes on a smudgy eosinophilic
appearance called fibrinoid necrosis, caused by protein
deposition
However, chronic immune complex disease develops
when there is persistent antigenemia or repeated exposure
to an antigen.
This occurs in some human diseases, such as systemic
lupus erythematosus (SLE).
Local Immune Complex Disease
Arthus reaction:
In which an area of tissue necrosis appears as a result of
acute immune complex vasculitis.
The reaction is produced experimentally by (injecting an
antigen into the skin of a previously immunized animal
Unit 4: Diseases of the Immune System
61
i.e., pre-formed antibodies against the antigen are already
present in the circulation).
Because of the initial antibody excess, immune complexes
are formed as the antigen diffuses into the vascular wall;
these are precipitated at the site of injection and trigger
the same inflammatory reaction and histologic appearance
as in systemic immune complex disease.
Arthus lesions evolve over a few hours and reach a peak 4
to 10 hours after injection, when the injection site
develops visible edema with severe hemorrhage,
occasionally followed by ulceration.
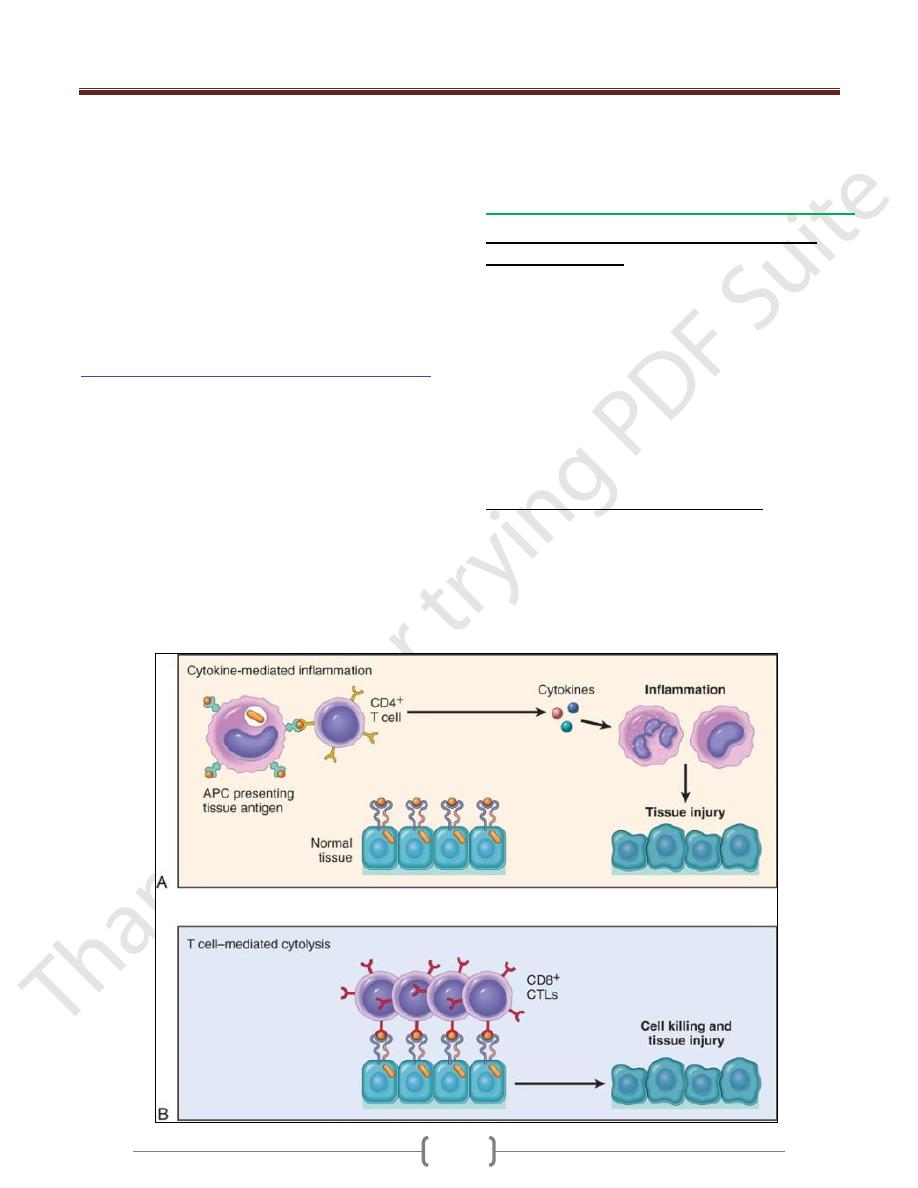
T cell–mediated (type IV) hypersensitivity
Are caused mainly by immune responses in which T
lymphocytes of the T
H
1 and T
H
17 subsets produce
cytokines that induce inflammation and activate
neutrophils and macrophages, which are responsible for
tissue injury. CD8+ CTLs also may contribute to injury
by directly killing host cells.
Two types of T cell reactions are capable of causing tissue
injury and disease:
(1) cytokine-mediated inflammation, in which the
cytokines are produced mainly by CD4+ T cells
(2) direct cell cytotoxicity, mediated by CD8+ Tc cells
In inflammation, exemplified by the delayed-type
hypersensitivity (DTH) reaction, CD4+ T cells of the T
H
1
and T
H
17 subsets secrete cytokines, which recruit and
activate other cells, especially macrophages, and these are
the major effector cells of injury.
Inflammatory reactions elicited by CD4+ T cells
1) First exposure to antigen and is essentially cell-
mediated immunity
Naive CD4+ T lymphocytes recognize peptide antigens of
self or microbial proteins in association with class II
MHC molecules on the surface of DCs (or macrophages)
that have processed the antigens.
If the DCs produce IL-12, the naive T cells differentiate
into effector cells of the T
H
1 type.
The cytokine IFN-γ, made by NK cells and by the T
H
1
cells themselves, further promotes T
H
1 differentiation,
providing a powerful positive feedback loop.
If the APCs produce IL-1, IL-6, or IL-23 instead of IL-
12, the CD4+ cells develop into T
H
17 effectors.
2) On subsequent exposure to the antigen,
The previously generated effector cells are recruited to the
site of antigen exposure and are activated by the antigen
presented by local APCs.
The T
H
1 cells secrete IFN-γ, which is the most potent
macrophage-activating cytokine known.
Activated macrophages also express more class II MHC
Unit 4: Diseases of the Immune System
62
molecules and costimulators, and the cells secrete more
IL-12, thus stimulating more T
H
1 responses.
T
H
17 effector cells secrete IL-17 which promote the
recruitment of neutrophils (and monocytes) and thus
induce inflammation. Because the cytokines produced by
the T cells enhance leukocyte recruitment and activation,
these inflammatory reactions become chronic unless the
offending agent is eliminated or the cycle is interrupted
therapeutically.
Contact dermatitis is an example of tissue injury resulting
from T cell-mediated inflammation. It is evoked by
contact with the active component of poison ivy and
poison oak, which probably becomes antigenic by binding
to a host protein).
On reexposure of a previously exposed person to the
plants, sensitized T
H
1 CD4+ cells accumulate in the
dermis and migrate toward the antigen within the
epidermis. Here they release cytokines that damage
keratinocytes, causing separation of these cells and
formation of an intraepidermal vesicle, and inflammation
manifested as a vesicular dermatitis.
type 1 diabetes
multiple sclerosis, are caused by T
H
1 and T
H
17 reactions
against self-antigens, and
Crohn disease may be caused by uncontrolled reactions
involving the same T cells but directed against intestinal
bacteria.
rejection of transplants
Delayed-Type Hypersensitivity
DTH is a T cell-mediated reaction that develops in
response to antigen challenge in a previously sensitized
individual. In contrast with immediate hypersensitivity,
the DTH reaction is delayed for 12 to 48 hours, which is
the time it takes for effector T cells to be recruited to the
site of antigen challenge and to be activated to secrete
cytokines.
Tuberculin reaction, elicited by challenge with a protein
extract of M. tuberculosis (tuberculin) in a person who has
previously been exposed to the tubercle bacillus. Between
8 and 12 hours after intracutaneous injection of
tuberculin, a local area of erythema and induration
appears, reaching a peak (typically 1 to 2 cm in diameter)
in 24 to 72 hours and thereafter slowly subsiding.
On histologic examination, the DTH reaction is
characterized by perivascular accumulation ("cuffing") of
CD4+ helper T cells and macrophages
Local secretion of cytokines by these cells leads to
increased microvascular permeability, giving rise to
dermal edema and fibrin deposition; the latter is the main
cause of the tissue induration in these responses.
DTH reactions are mediated primarily by T
H
1 cells; the
contribution of T
H
17 cells is unclear.
The tuberculin response is used to screen populations for
people who have had previous exposure to tuberculosis
and therefore have circulating memory T cells specific for
mycobacterial proteins.
Notably, immunosuppression or loss of CD4+ T cells
(e.g., resulting from HIV infection) may lead to a
negative tuberculin response even in the presence of a
severe infection.
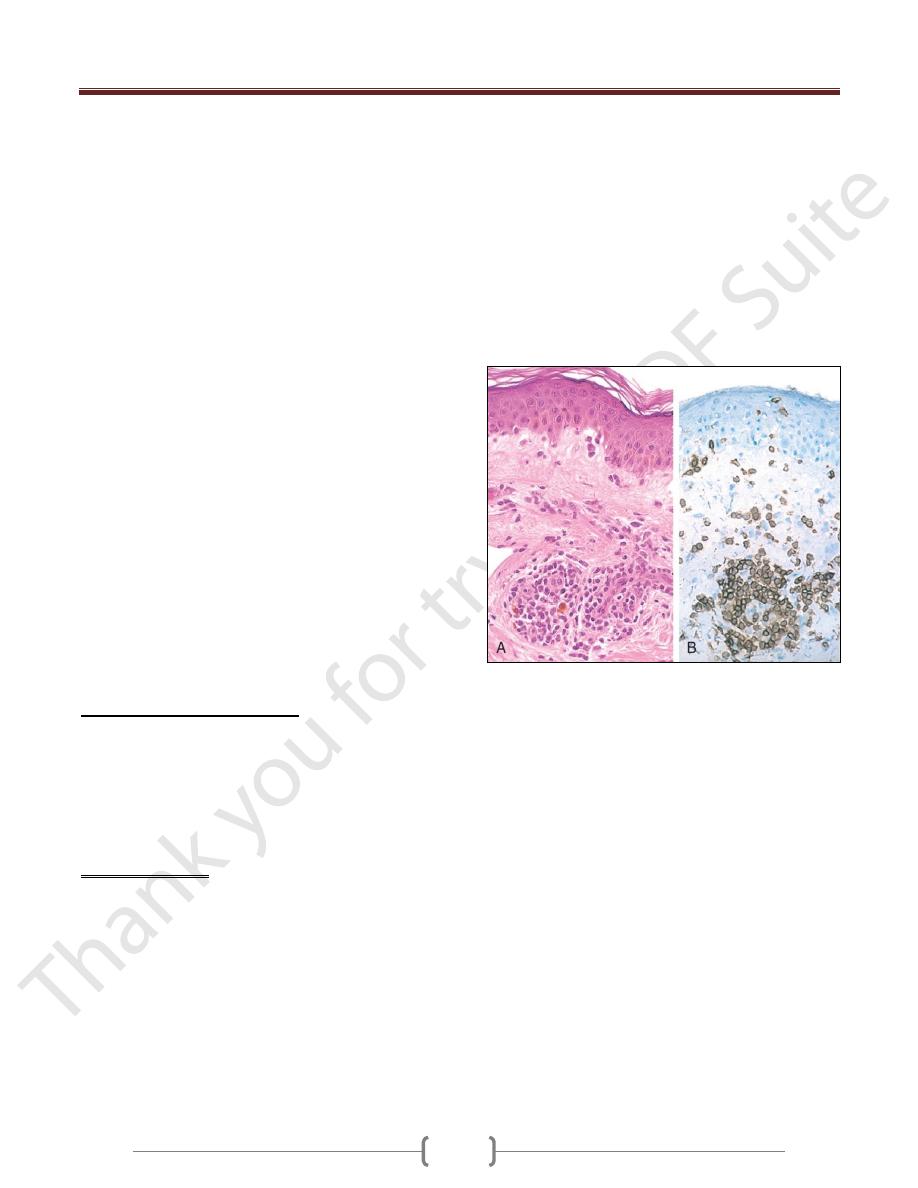
Delayed-type hypersensitivity reaction in the skin. A,
Perivascular accumulation ("cuffing") of mononuclear
inflammatory cells (lymphocytes and macrophages), with
associated dermal edema and fibrin deposition. B,
Immunoperoxidase staining reveals a predominantly perivascular
cellular infiltrate that marks positively with anti-CD4 antibodies
Prolonged DTH reactions against persistent microbes or
other stimuli may result in a special morphologic pattern
of reaction called granulomatous inflammation.
The initial perivascular CD4+ T cell infiltrate is
progressively replaced by macrophages over a period of 2
to 3 weeks. They become large, flat, and eosinophilic, and
are called epithelioid cells.
The epithelioid cells occasionally fuse under the influence
of cytokines (e.g., IFN-γ) to form multinucleate giant cells.
A microscopic aggregate of epithelioid cells, typically
surrounded by a collar of lymphocytes, is called a granuloma
The process is essentially a chronic form of T
H
1-mediated
inflammation and macrophage activation
Older granulomas develop an enclosing rim of fibroblasts
and connective tissue.
Unit 4: Diseases of the Immune System
63
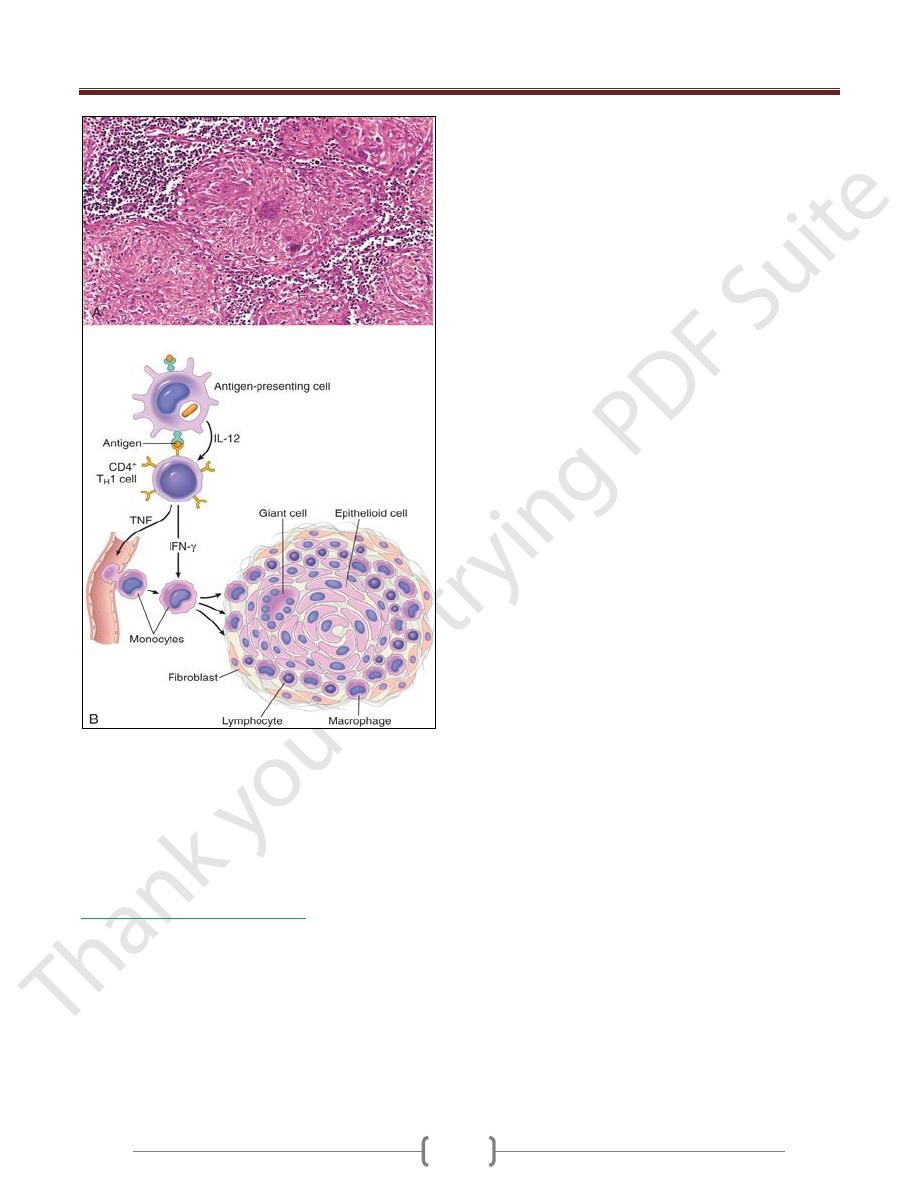
Granulomatous inflammation.
A, A section of a lymph node shows several granulomas, each
made up of an aggregate of epithelioid cells and surrounded by
lymphocytes. The granuloma in the center shows several
multinucleate giant cells.
B, The events that give rise to the formation of granulomas in
type IV hypersensitivity reactions. Note the role played by T-
cell-derived cytokines.
T Cell-Mediated Cytotoxicity
CD8+ CTLs kill antigen-bearing target cells.
Class I MHC molecules bind to intracellular peptide
antigens and present the peptides to CD8+ T lymphocytes,
stimulating the differentiation of these T cells into effector
cells called CTLs.
CTLs play a critical role in resistance to virus infections
and some tumors.
The principal mechanism of killing by CTLs is dependent
on the perforin-granzyme system. Perforin and granzymes
are stored in the granules of CTLs and are rapidly released
when CTLs engage their targets.
Perforin binds to the plasma membrane of the target cells
and promotes the entry of granzymes, which are proteases
that specifically cleave and thereby activate cellular
caspases. These enzymes induce apoptotic death of the
target cells
CTLs play an important role in the rejection of solid-
organ transplants and may contribute to many
immunologic diseases, such as type 1 diabetes (in which
insulin-producing β cells in pancreatic islets are destroyed
by an autoimmune T cell reaction).
CD8+ T cells may also secrete IFN-γ and contribute to
cytokine-mediated inflammation, but less so than CD4+
cells.
Unit 4: Diseases of the Immune System
64
Autoimmune diseases
Is an immune reaction to self-antigens (i.e., autoimmunity
due to presence of autoreactive antibodies or T cells
Presumed autoimmune diseases range from those in
which specific immune responses are directed against one
particular organ or cell type and result in localized
tissue damage, to multisystem diseases characterized by
lesions in many organs and associated with multiple
autoantibodies or cell-mediated reactions against
numerous self-antigens
In the systemic diseases, the lesions affect principally the
connective tissue and blood vessels of the various
organs involved.
The diseases are often referred to as "collagen vascular"
or "connective tissue" disorders.
Immunological Tolerance
lack of immune responsiveness to one's own tissue antigens
Central tolerance. This refers to deletion of self-reactive
T and B lymphocytes during their maturation in central
lymphoid organs (i.e., in the thymus for T cells and in the
bone marrow for B cells).
Peripheral tolerance. Self-reactive T cells that escape
negative selection in the thymus can potentially dangerous
unless they are deleted.
Several mechanisms in the peripheral tissues that
silence autoreactive T cells have been identified:
1) Anergy: This refers to functional inactivation (rather than
death) of lymphocytes induced by encounter with antigens
under certain conditions
2) Suppression by regulatory T cells: The responses of T
lymphocytes to self-antigens may be actively suppressed
by regulatory T cells (Treg).
3) Activation-induced cell death: apoptosis of mature
lymphocytes as a result of self-antigen recognition.
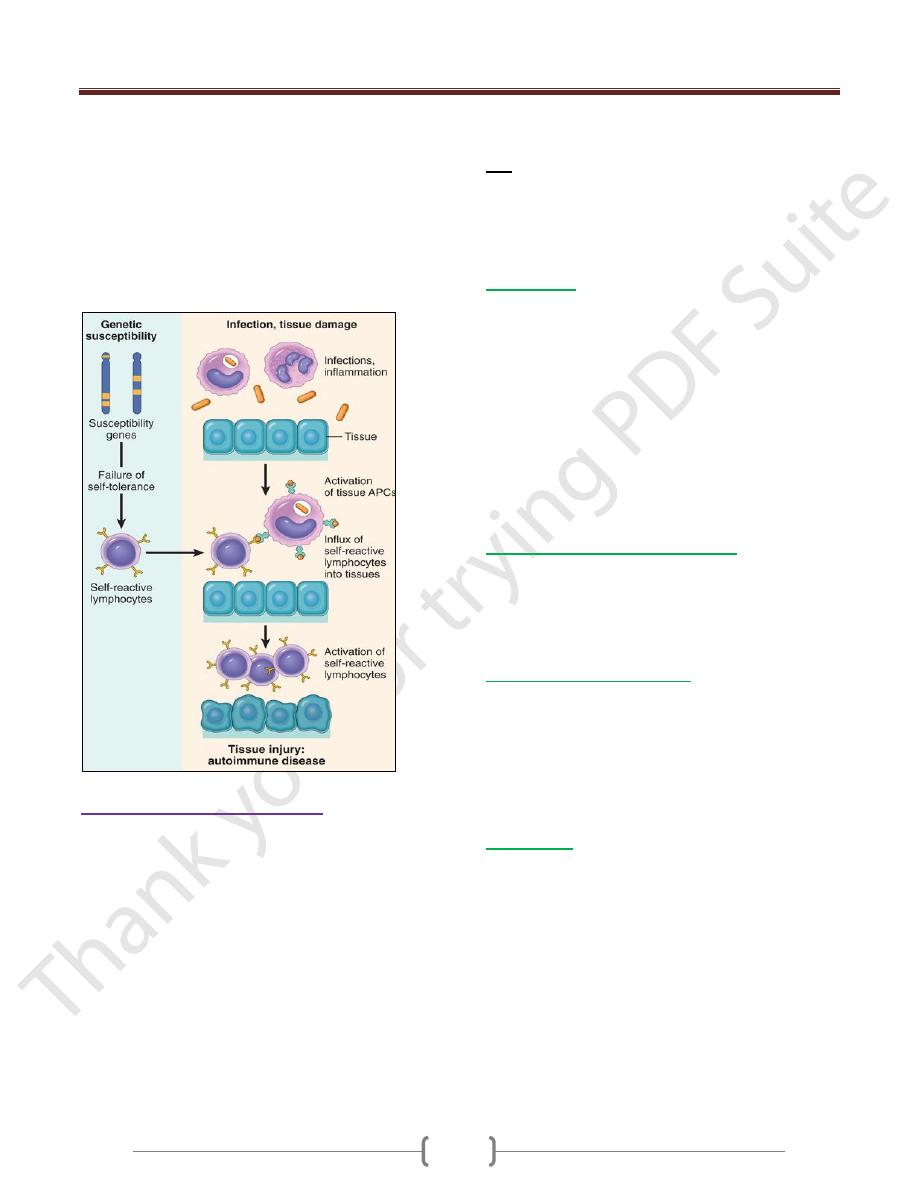
Mechanisms of Autoimmunity
The breakdown of self-tolerance and the development of
autoimmunity are probably related to the inheritance of
various susceptibility genes and environmental factors
that cause changes in tissues, often induced by infections or
injury, that alter the display & recognition of self-antigens
Genetic Factors in Autoimmunity
Autoimmune diseases have a tendency to run in families,
and there is a greater incidence of the same disease in
monozygotic than in dizygotic twins
Several autoimmune diseases are linked with the HLA
locus, especially class II alleles (HLA-DR, -DQ).
Genome-wide linkage analyses are revealing many
genetic loci that are associated with different
autoimmune diseases due to mutation or polymorphism
Role of Infections and Tissue Injury
Viruses and other microbes may share cross-reacting
epitopes with self-antigens,
Microbial infections with resultant tissue necrosis and
inflammation can cause upregulation of costimulatory
molecules on APCs in the tissue, thus favoring a break-
Central and
peripheral
tolerance (T -and
B- cell)
Unit 4: Diseases of the Immune System
65
down of T cell anergy and subsequent T cell activation.
Tissue antigens may be altered by a variety of
environmental insults, like ultraviolet (UV) radiation
causes cell death and may lead to the exposure of nuclear
antigens, which elicit pathologic immune responses
in lupus; this mechanism is the proposed explanation for
the association of lupus flares with exposure to
sunlight. Smoking had an effect
there is a strong gender bias of autoimmunity,♀>♂
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a multisystem
autoimmune disease of variable clinical behavior.
Clinically, it is an unpredictable, remitting and relapsing
disease of acute or insidious onset that may involve
virtually any organ in the body
It affects principally the skin, kidneys, serosal
membranes, joints, and heart.
Immunologically, the disease is associated with an
enormous array of autoantibodies, classically including
antinuclear antibodies (ANAs). The clinical presentation
of SLE is so variable, with so many overlapping features
with those of other autoimmune diseases (RA,
polymyositis, and others), that it has been necessary to
develop diagnostic criteria for SLE
The diagnosis is established by demonstration of 4 or
more of the criteria during any interval of observation.
4/11
1. Malar rash 2. Discoid rash 3. Photosensitivity
4. Oral ulcers 5. Arthritis 6. Serositis 7. Renal disorder
8. Neurologic disorder 9. Hematologic disorder
10. Immunologic disorder 11. Antinuclear antibody
Pathogenesis
1) Genetic Factors. Many lines of evidence support a
genetic predisposition to SLE.(Familial association,
HLA association, Genetic deficiencies of classical
pathway complement proteins, especially C1q, C2, or C4,
are seen in about 10% of patients with SLE.)
2) Environmental Factors.(Ultraviolet (UV) radiation,
Cigarette smoking, Sex hormones, Drugs
3) Immunologic Abnormalities in SLE.
Production abnormally large amounts of IFN-α.
TLR signals (Toll -like receptors) that activate B cells
specific for self-nuclear Ags. Failure of B cell tolerance
Spectrum of Autoantibodies in SLE
Antibodies have been identified against a host of nuclear &
cytoplasmic components of the cell(DNA, histones, nucleolar
Another group of antibodies is directed against surface
Ags of blood cells, platelets, lymphocytes, phospholipids
Mechanisms of Tissue Injury
Most organ damage in SLE is caused by immune complex
deposition. (Type III hypersensitivity)
Autoantibodies of different specificities contribute to the
pathology & clinical manifestations of SLE (type II hyper-
sensitivity). Autoantibodies against red cells, white cells, &
platelets opsonize these cells and lead to their phagocytosis
Morphology
The morphologic changes in SLE are therefore extremely
variable and depend on the nature of the autoantibodies,
the tissue in which immune complexes deposit, and the
course and duration of disease. The most characteristic
morphologic changes result from the deposition of
immune complexes in a variety of tissues.
1) Blood Vessels. An acute necrotizing vasculitis
2) Kidneys. The pathogenesis of all forms
of glomerulonephritis in SLE involves deposition of
DNA-anti-DNA complexes within the glomeruli.
3) Skin is involved in a majority of patients; a characteristic
erythematous or maculopapular eruption over the malar
eminences and bridge of the nose ("butterfly pattern")
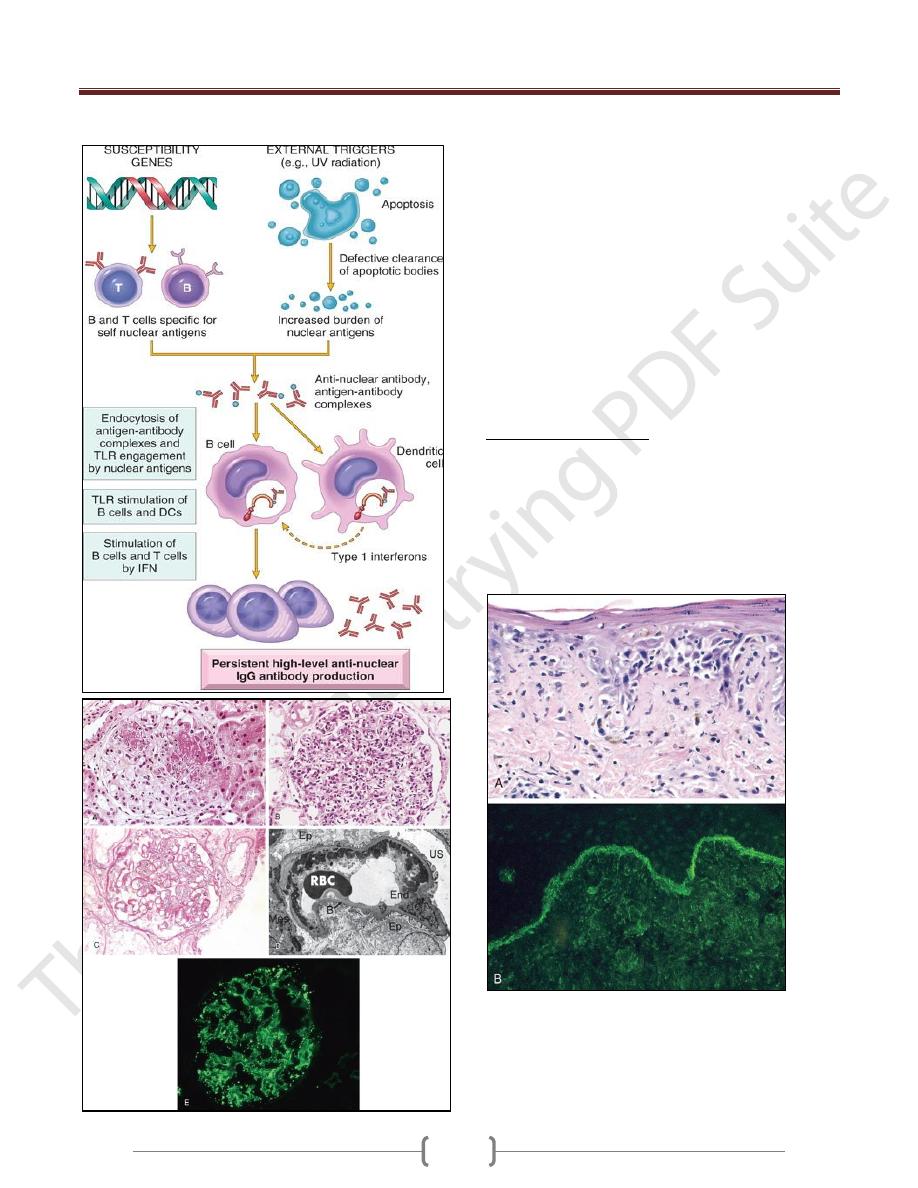
Unit 4: Diseases of the Immune System
66
4) Joints. 5) CNS. 6) spleen 7) heart 8) liver
Lupus nephritis
A. Focal lupus nephritis, with two necrotizing lesions in a glomerulus
(segmental distribution) (H&E stain).
B. Diffuse lupus nephritis. Note the marked global increase in
cellularity throughout the glomerulus (H&E stain).
C. Lupus nephritis showing a glomerulus with several "wire loop"
lesions representing extensive subendothelial deposits of immune
complexes (periodic acid Schiff stain).
D. Electron micrograph of a renal glomerular capillary loop from a
patient with SLE nephritis. Confluent subendothelial dense deposits
correspond to "wire loops" seen by light microscopy.
E. Deposition of IgG antibody in a granular pattern, detected by
immunofluorescence.
B, basement membrane; End, endothelium;
Ep, epithelial cell with foot processes; Mes, mesangium; RBC, red
blood cell in capillary lumen; US, urinary space; *, electron-dense
deposits in subendothelial location.
Clinical Manifestations
The patient is a young woman with some, but rarely all, of
the following features: a butterfly rash over the face,
fever, pain and swelling in one or more peripheral joints
(hands and wrists, knees, feet, ankles, elbows, shoulders),
pleuritic chest pain, and photosensitivity.
The most common causes of death are renal failure,
intercurrent infections, and cardiovascular disease
Systemic lupus erythematosus involving the skin.
A, An H&E-stained section shows liquefactive degeneration of the
basal layer of the epidermis and edema at the dermoepidermal
junction.
B, An immunofluorescence micrograph stained for IgG reveals
deposits of immunoglobulin along the dermoepidermal junction.
H&E, hematoxylin-eosin; IgG, immunoglobulin G.

Unit 4: Diseases of the Immune System
67
Rheumatoid Arthritis (RA)
Is a systemic, chronic inflammatory disease affecting
many tissues but principally attacking the joints to
produce a nonsuppurative proliferative synovitis that
frequently progresses to destroy articular cartilage and
underlying bone with resulting disabling arthritis.
Morphology
RA typically presents as symmetric arthritis,
principally affecting the small joints of the hands and
feet, ankles, knees, wrists, elbows, and shoulders.
Typically, the proximal interphalangeal and
metacarpophalangeal joints are affected, but distal
interphalangeal joints are spared.
Histologically, the affected joints show chronic
synovitis, characterized by
1) synovial cell hyperplasia and proliferation;
2) dense perivascular inflammatory cell infiltrates
(frequently forming lymphoid follicles) in the synovium
composed of CD4+ T cells, plasma cells & macrophages;
3) increased vascularity due to angiogenesis;
4) neutrophils and aggregates of organizing fibrin on the
synovial surface and in the joint space; and
5) increased osteoclast activity in the underlying bone,
leading to synovial penetration and bone erosion
The classic appearance is that of a pannus, formed by
proliferating synovial-lining cells admixed with
inflammatory cells, granulation tissue, and fibrous
connective tissue; the overgrowth of this tissue is so
exuberant that the usually thin, smooth synovial
membrane is transformed into , edematous, frondlike
(villous) projections
With full-blown inflammatory joint involvement,
periarticular soft tissue edema usually develops,
classically manifested first by fusiform swelling of the
proximal interphalangeal joints
With progression of the disease, the articular cartilage
subjacent to the pannus is eroded and, virtually destroyed.
The subarticular bone may also be attacked and eroded.
Eventually the pannus fills the joint space, and subsequent
fibrosis and calcification may cause permanent ankylosis.
Rheumatoid arthritis. A, A joint lesion. B, Low magnification reveals
marked synovial hypertrophy with formation of villi. C, At higher
magnification, dense lymphoid aggregates are seen in the synovium
Sjögren Syndrome
Sjögren syndrome is a clinicopathologic entity
characterized by dry eyes (keratoconjunctivitis sicca)
and dry mouth (xerostomia), resulting from immune-
mediated destruction of the lacrimal and salivary glands.
It occurs as an isolated disorder (primary form), or more
often in association with another autoimmune disease
(secondary form).

Unit 4: Diseases of the Immune System
68
Involved tissues show an intense lymphocyte (primarily
activated CD4+ T cells) & plasma-cell infiltrate,
occasionally forming lymphoid follicles with germinal
centers.
The disease is believed to be caused by an autoimmune
T-cell reaction against an unknown self-antigen(s)
expressed in these glands, or immune reactions against the
antigens of a virus that infects the tissues The disease is
believed to be caused by an autoimmune T-cell reaction
against an unknown self-antigen(s) expressed in these
glands, or immune reactions against the antigens of a
virus that infects the tissues
Sjögren syndrome. A, Enlargement of the salivary gland. B,
The histologic view shows intense lymphocytic and plasma cell
infiltration with ductal epithelial hyperplasia.
Systemic Sclerosis (Scleroderma)
This disorder is better labeled (SS), because it is
characterized by excessive fibrosis throughout the body
and not just the skin.
Cutaneous involvement is the usual presenting symptom
in approximately 95% of cases,
but it is the visceral involvement-of the gastrointestinal
tract, lungs, kidneys, heart, and skeletal muscles-that
produces the major morbidity and mortality.
Fibroblast activation with excessive fibrosis is the
hallmark of systemic sclerosis. .
It is proposed that CD4+ cells responding to an
unidentified antigen accumulate in the skin and release
cytokines that activate mast cells and macrophages; in
turn, these cells release fibrogenic cytokines such as IL-
1, PDGF, TGF-β, and fibroblast growth factors.
B-cell activation also occurs, as indicated by the presence
of hypergammaglobulinemia and ANAs.
Almost all patients develop Raynaud phenomenon
Endothelial injury and microvascular disease are
commonly present in the lesions of systemic sclerosis
Systemic sclerosis.
A, Normal skin.
B, Extensive deposition of dense collagen in the dermis. C, The
extensive subcutaneous fibrosis has virtually immobilized the
fingers, creating a clawlike flexion deformity.
Loss of blood supply has led to cutaneous ulcerations.
Pathogenesis of SS: unknown
external stimuli cause vascular
abnormalities and immune
activation in genetically
susceptible individual leads to
excessive fibrosis

Unit 4: Diseases of the Immune System
69
Inflammatory Myopathies
Inflammatory myopathies make up a heterogeneous group
of rare disorders characterized by immune-mediated
muscle injury and inflammation.
Three disorders-:
polymyositis,
dermatomyositis,
and inclusion body myositis-have been described.
Women with dermatomyositis have a slightly increased risk
of developing visceral cancers (of the lung, ovary, stomach)
The immunologic evidence supports antibody-mediated
tissue injury in dermatomyositis, whereas polymyositis and
inclusion body myositis seem to be mediated by CTLs.
ANAs are present in most patients.
Only Jo-1 antibodies, directed against transfer RNA
synthetase, are specific for this group of disorders
Rejection of transplants
The major barrier to transplantation of organs from one
individual to another of the same species (called allografts)
is immunologic rejection of the transplanted tissue
Rejection of allografts is a response to MHC molecules,
which are so polymorphic that no two individuals in an
outbred population are likely to express exactly the same
set of MHC molecules
Effector Mechanisms of Graft Rejection
Recognition of alloAgs by direct and indirect pathway
lead to T cells and antibodies reactive with the graft are
involved in the rejection of most solid-organ allografts
Morphology (Types)
Hyperacute Rejection
.
Occurs within minutes to a few hours after transplantation
in a presensitized host and is typically recognized by the
surgeon just after the vascular anastomosis is completed.
The histology is characterized by widespread acute
arteritis and arteriolitis, vessel thrombosis, and
ischemic necrosis, all resulting
From the binding of preformed antibodies to graft
endothelium. Virtually all arterioles and arteries exhibit
characteristic acute fibrinoid necrosis of their walls,
Acute cellular rejection
Is most commonly seen within the first months after
transplantation and is typically accompanied by clinical
signs of renal failure.
Histologically, there is usually extensive interstitial CD4+
and CD8+ T-cell infiltration with edema and mild
interstitial hemorrhage.
Glomerular and peritubular capillaries contain large
numbers of mononuclear cells, which may also invade
the tubules and cause focal tubular necrosis. In addition to
tubular injury, CD8+ T cells may also injure the
endothelium, causing an endotheliitis.
Acute humoral rejection (rejection vasculitis)
caused by antibodies
The histologic lesions may take the form of necrotizing
vasculitis with endothelial cell necrosis; neutrophilic
infiltration; deposition of antibody, complement, and
fibrin; and thrombosis.
Such lesions may be associated with ischemic necrosis of
the renal parenchyma.
The resultant narrowing of the arterioles may cause
infarction or renal cortical atrophy.
The proliferative vascular lesions mimic arteriosclerotic
thickening and are believed to be caused by cytokines
Chronic Rejection
:
Present clinically late after transplantation (months to
years) with a progressive rise in serum creatinine levels
(an index of renal dysfunction) over a period of 4 to 6
months.
The vascular changes occur predominantly in the arteries
and arterioles, which exhibit intimal smooth muscle cell
proliferation and extracellular matrix synthesis
Result in renal ischemia manifested by loss or hyalinization
of glomeruli, interstitial fibrosis, and tubular atrophy.
The vascular lesion may be caused by cytokines released by
activated T cells that act on the cells of the vascular wall.
Chronic rejection does not respond to standard immune-
suppression regimens& need other renal transplant
Hyperacute Rejection. Hyperacute rejection occurs
within minutes to a few hours after transplantation in a
presensitized host and is typically recognized by the
surgeon just after the vascular anastomosis is completed.

Unit 4: Diseases of the Immune System
70
Morphologic patterns of graft rejection.
A, Hyperacute rejection of a kidney allograft showing endothelial
damage, platelet and thrombi in a glomerulus.
B, Acute cellular rejection of a kidney allograft with inflammatory
cells in the interstitium and between epithelial cells of the tubules.
C, Acute humoral rejection of a kidney allograft (rejection
vasculitis) with inflammatory cells & proliferating smooth muscle
cells in the intima
D, Chronic rejection in a kidney allograft with graft arteriosclerosis.
The arterial lumen is replaced by an accumulation of smooth muscle
cells and connective tissue in the intima.
Transplantation of Hematopoietic Cells
Bone marrow transplantation is increasingly used as
therapy for hematopoietic and some nonhematopoietic
malignancies, aplastic anemias, and certain immune
deficiency states
HSC previously was obtained from donor bone marrow,
now was harvested from peripheral blood after
administration of hematopoietic growth factor or from the
umbilical cord of newborn as a rich source of HSC
Two complications were raised from these
transplantations:
1) 1-Graft –versus –host Disease (GVHD) occurs when
immunologically competent T cells (or their precursors)
are transplanted into recipients who are immunologically
compromised. Either acute or chronic
2) 2-Immune deficiency leads to viral infections (EBV, CMV)

Unit 4: Diseases of the Immune System
71
Immune deficiency diseases
Immune deficiency diseases may be caused by inherited
defects affecting immune system development, or they
may result from secondary effects of other diseases (e.g.,
infection, malnutrition, aging, immunosuppression,
autoimmunity, or chemotherapy).
Clinically, patients with immune deficiency present with
increased susceptibility to infections as well as to certain
forms of cancer.
Primary Immune Deficiencies (congenital)
Most primary immune deficiency diseases are genetically
determined and affect either adaptive immunity (i.e.,
humoral or cellular) or innate host defense mechanisms,
including complement proteins and cells such as
phagocytes and NK cells.
Most primary immune deficiencies occur early in life
(between 6 months and 2 years of age).
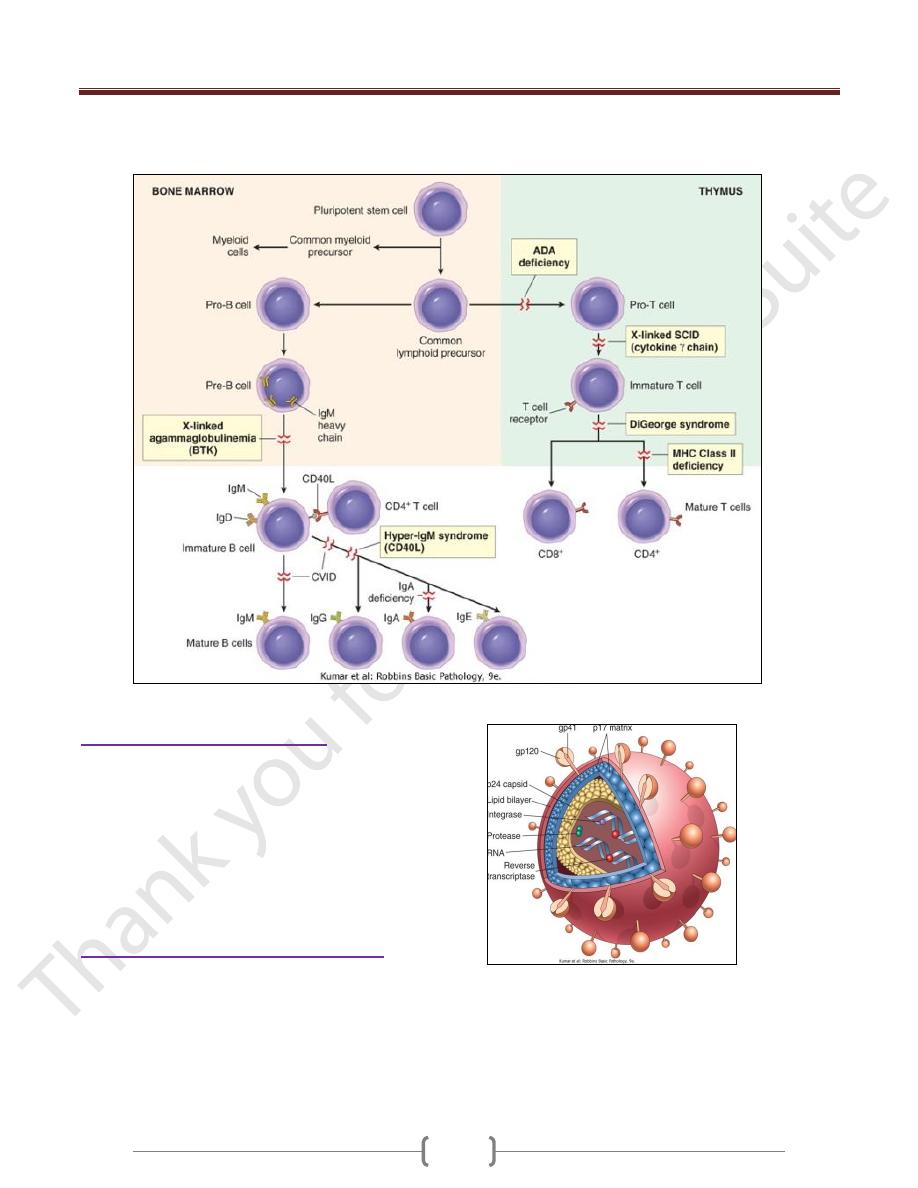
X-Linked Agammaglobulinemia (XLA, Bruton
Disease)
It is characterized by the failure of pre-B cells to
differentiate into B cells; as a consequence, and as the
name implies, there is a resultant absence of gamma
globulin in the blood.
B-cell maturation stops because of mutations in a tyrosine
kinase that is associated with pre-B-cell signal transduction
Absent or markedly decreased numbers of B cells in the
circulation, depressed serum levels of all classes of
immunoglobulins.
The numbers of pre-B cells in the bone marrow may be
normal or reduced.
Underdeveloped germinal centers in peripheral lymphoid
tissues, Absence of plasma cells throughout the body
Normal T-cell-mediated responses
Common Variable Immunodeficiency
This is a heterogeneous group of disorders characterized by:
hypogammaglobulinemia, impaired Ab responses to
infection (or vaccination), increased susceptibility to
infections
The basis of the Ig deficiency is variable (hence the name).
Isolated IgA Deficiency
The pathogenesis of IgA deficiency seems to involve a
block in the terminal differentiation of IgA-secreting B
cells to plasma cells; IgM and IgG subclasses of antibodies
are present in normal or even supranormal levels
Hyper-IgM Syndrome
Patients with the hyper-IgM syndrome produce normal (or
even supranormal) levels of IgM antibodies to antigens
but lack the ability to produce the IgG, IgA, or IgE
isotypes; the underlying defect is an inability of T cells
to induce B-cell isotype switching
The most common genetic abnormality is mutation of the
gene encoding CD40L.
The contact-dependent signals are provided by interaction
between CD40 molecules on B cells and CD40L (also
known as CD154), expressed on activated helper T cells.
There is also a defect in cell-mediated immunity because
the CD40-CD40L interaction is critical for helper T cell-
mediated activation of macrophages, the central reaction
of cell-mediated immunity.
These patients are also susceptible to a variety of
intracellular pathogens that are normally combated by
cell-mediated immunity
Thymic Hypoplasia: DiGeorge Syndrome
The disorder is a consequence of a developmental
malformation affecting the third and fourth pharyngeal
pouches-structures that give rise to the thymus,
parathyroid glands, and portions of the face and aortic
arch. Thus, in addition to the thymic and T-cell defects,
there may be parathyroid gland hypoplasia resulting in
hypocalcemic tetany,
Severe Combined Immunodeficiency (SCID)
Defects in both humoral & cell-mediated immune responses
Affected infants are susceptible to severe recurrent
infections by a wide array of pathogens, including
bacteria, viruses, fungi, and protozoans; opportunistic
infections by Candida.
These are caused by mutations in the gene encoding the
common γ chain shared by the receptors for the cytokines
IL-2, IL-4, IL-7, IL-9, and IL-15.
IL-7 is the most important in this disease because it is the
growth factor responsible for stimulating the survival
and expansion of immature B- and T-cell precursors in
the generative lymphoid organs.
It is X- linked (X-SCID) caused by mutations in
adenosine deaminase (ADA), an enzyme involved in
purine metabolism. ADA deficiency results in
accumulation of adenosine, which inhibit DNA synthesis
& are toxic to lymphocytes. It is Autosomal SCID
Patients with SCID are currently treated by bone marrow
transplantation.
Unit 4: Diseases of the Immune System
72
X-SCID is the first disease in which gene therapy has
been used to successfully replace the mutated gene
Secondary Immune Deficiencies
Immune deficiencies secondary to other diseases or
therapies are much more common than the primary
(inherited) disorders.
Secondary immune deficiencies may be encountered in
patients with malnutrition, infection, cancer, renal
disease, or sarcoidosis. However, the most common cases
of immune deficiency are therapy-induced suppression of
the bone marrow and of lymphocyte function.
Acquired Immunodeficiency Syndrome
AIDS is a retroviral disease caused by the human
immunodeficiency virus (HIV). It is characterized by
infection and depletion of CD4+ T lymphocytes, and by
profound immunosuppression leading to opportunistic
infections, secondary neoplasms, and neurologic
manifestations

Unit 4: Diseases of the Immune System
73
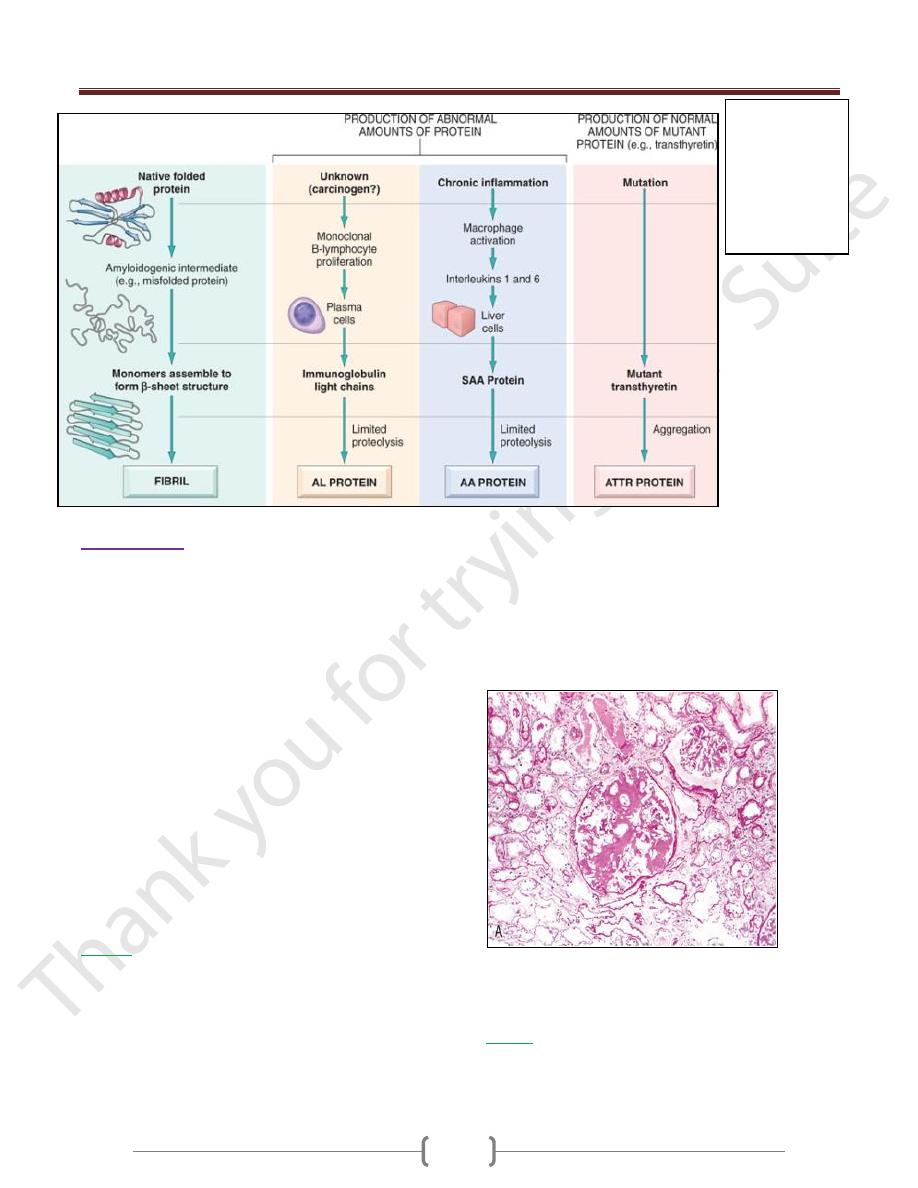
Amyloidosis
Is a condition associated with a number of inherited and
inflammatory disorders in which extracellular deposits
of fibrillar proteins are responsible for tissue damage
and functional compromise
These abnormal fibrils are produced by the aggregation
of misfolded proteins
The presence of abundant charged sugar groups in these
adsorbed proteins give the deposits staining characteristics
that were thought to resemble starch (amylose).
Therefore, the deposits were called amyloid, despite the
realization that the deposits are unrelated to "starch."
Pathogenesis of Amyloid Deposition
Amyloidosis is fundamentally a disorder of protein
misfolding.
More than 20 (at last count, 23) different proteins can
aggregate & form fibrils with the appearance of amyloid.
All amyloid deposits are composed of nonbranching
fibrils that are wound together .The dye Congo red
binds to these fibrils and produces a red-green
Structure of amyloid.
A, Schematic diagram of an amyloid fiber showing fibrils (4
shown, may be up to 6) wound around one another with
regularly spaced binding of the Congo red dye.
B, Congo red staining shows an apple-green birefringence under
polarized light, a diagnostic feature of amyloid.
C, Electron micrograph of 7.5-10 nm amyloid fibrils.
The proteins that form amyloid fall into 2 general
categories:
1) normal proteins that have an inherent tendency to fold
improperly,
2) Mutant proteins that is prone to misfolding and
subsequent aggregation. three are most common:
The AL (amyloid light chain) protein: is produced by
plasma cells and is made up of complete immunoglobulin
light chains,
The AA (amyloid-associated) fibril: is a protein derived
from serum precursor called SAA (serum amyloid-
associated) protein that is synthesized in the liver under
the influence of cytokines such as IL-6 and IL-1 that are
produced during inflammation.
Aβ amyloid: is found in cerebral lesion of Alzheimer
disease . constitutes the core of cerebral plaques
Transthyretin (TTR) : s a normal serum protein that
binds and transports thyroxine and retinol,
β
2
-Microglobulin: a component of MHC class I molecules
Classification of Amyloidosis
Amyloid may be
systemic (generalized),
involving
several organ systems,
On clinical grounds, the systemic, or generalized, pattern
is subclassified into primary amyloidosis when
associated with a monoclonal plasma cell proliferation &
Secondary amyloidosis when it occurs as a complication
of an underlying chronic inflammatory
Or it may be
localized
, when deposits are limited to a
single organ, such as the heart.
Hereditary or familial amyloidosis constitutes a
separate, heterogeneous group, with several distinctive
patterns of organ involvement.
Unit 4: Diseases of the Immune System
74
Morphology
There are no consistent or distinctive patterns of organ or
tissue distribution of amyloid deposits
In amyloidosis secondary to chronic inflammatory
disorders, kidneys, liver, spleen, lymph nodes, adrenals,
and thyroid, as well as many other tissues,
On histologic examination, the amyloid deposition is
always extracellular and begins between cells, often
closely adjacent to basement membranes. As the amyloid
accumulates, it surrounding the cells and destroying them.
In the AL form, perivascular and vascular localizations
are common.
The histologic diagnosis of amyloid is based almost
entirely on its staining characteristics. The most
commonly used staining technique uses the dye Congo
red, which under ordinary light imparts a pink or red
color to amyloid deposits. Under polarized light the
Congo red-stained amyloid shows so-called apple-
green birefringence
Kidney
Grossly, the kidney may appear unchanged, or it may be
abnormally large, pale, gray, and firm; in long-standing
cases, the kidney may be reduced in size.
Microscopically, the amyloid deposits are found
principally in the glomeruli, but they also are present in
the interstitial peritubular tissue as well as in the walls
of the blood vessels.
The glomerulus first develops focal deposits within the
mesangial matrix and diffuse or nodular thickenings
of the basement membranes of the capillary loops.
The interstitial peritubular deposits frequently are
associated with the appearance of amorphous pink casts
within the tubular lumens, presumably of a
proteinaceous nature.
Amyloidosis. A, Amyloidosis of the kidney. The glomerular
architecture is almost totally obliterated by the massive
accumulation of amyloid
Spleen
The deposits may be virtually limited to the splenic
follicles, splenic sinuses, splenic pulp, producing tapioca-
like granules
Pathogenesis of
amyloidosis. The
proposed
mechanisms
underlying
deposition of the
major forms of
amyloid fibrils
Unit 4: Diseases of the Immune System
75
Gross examination ("sago spleen"), the spleen is firm in
consistency.
The presence of blood in splenic sinuses usually imparts a
reddish color to the waxy, friable deposits.
Liver
Grossly: massive enlargement liver, is extremely pale,
grayish, and waxy on both the external surface and the cut
section.
Histologic analysis : amyloid deposits first appear in
the space of Disse and then progressively enlarge to
encroach on the adjacent hepatic parenchyma & sinusoids
The trapped liver cells undergo compression atrophy and
are eventually replaced by sheets of amyloid;
Amyloidosis.
A, A section of the liver stained with Congo red reveals pink-red
deposits of amyloid in the walls of blood vessels and along
sinusoids.
B, Note the yellow-green birefringence of the deposits when
observed by polarizing microscope
Heart
Grossly The deposits may not be evident on gross
examination, or they may cause minimal to moderate
cardiac enlargement. The most characteristic gross
findings are gray-pink, dewdrop-like subendocardial
elevations, particularly evident in the atrial chambers.
histologic examination, deposits typically are found
throughout the myocardium, beginning between myocardial
fibers and eventually causing their pressure atrophy
Cardiac amyloidosis. The atrophic myocardial fibers are
separated by structureless, pink-staining amyloid.
Clinical Course
The clinical course depends on organs affected and the
severity of the involvement.
Nonspecific complaints such as weakness, fatigue, and
weight loss are the most common presenting
manifestations.
Later in the course, amyloidosis tends to manifest in one
of several ways: renal disease, hepatomegaly,
splenomegaly, or cardiac abnormalities.
Diagnosis
Biopsy and subsequent Congo red staining is the most
important tool in the diagnosis of amyloidosis.
Renal biopsy is useful in the presence of urinary
abnormalities.
Rectal and gingival biopsy specimens contain amyloid in
as many as 75% of cases with generalized amyloidosis.
Mean survival time after diagnosis ranging from 1 to 3
years.
Patients with myeloma-associated amyloidosis have a
poorer prognosis, although they may respond to
cytotoxic drugs used to treat the underlying disorder.
Resorption of amyloid after treatment of the associated
condition has been reported, but this is a rare occurrence.