
34

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
35
Lecture 1 – Edema, Hyperemia and
congestion & Hemorrhage
Edema
Approximately 60% of lean body weight is water, two-
thirds of which is intracellular and the remainder is in
extracellular compartments, mostly as interstitial fluid;
only 5% of total body water is in blood plasma. The term
edema signifies increased fluid in the interstitial tissue
spaces; fluid collections in different body cavities are
variously designated hydrothorax, hydropericardium, or
hydroperitoneum (the last is more commonly called
ascites). Anasarca is a severe and generalized edema with
profound subcutaneous tissue swelling.
The movement of fluid between vascular and interstitial
spaces is controlled mainly by the opposing effects of
vascular hydrostatic pressure and plasma colloid osmotic
pressure. Normally, the exit of fluid into the interstitium
from the arteriolar end of the microcirculation is nearly
balanced by inflow at the venular end; the lymphatics
drain a small residual amount of excess interstitial fluid.
Either increased capillary pressure or diminished colloid
osmotic pressure can result in increased interstitial fluid
As extravascular fluid accumulates in either case, the
increased tissue hydrostatic and plasma osmotic pressures
eventually achieve a new equilibrium, and water re-enters
the venules. Excess interstitial edema fluid is removed by
lymphatic drainage, ultimately returning to the
bloodstream via the thoracic duct); clearly, lymphatic
obstruction (e.g., due to scarring or tumor) can also impair
fluid drainage and cause edema. Finally, sodium retention
(with its obligatory associated water) due to renal disease
can also cause edema.
The edema fluid occurring with volume or pressure
overload, or under conditions of reduced plasma protein,
is typically a protein-poor transudate; it has a specific
gravity less than 1.012. Conversely, because of the
increased vascular permeability, inflammatory edema is a
protein-rich exudate with a specific gravity that is usually
greater than 1.020
Increased Hydrostatic Pressure
Localized increases in intravascular pressure can result
from impaired venous return; for example, lower
extremity deep venous thrombosis can cause edema
restricted to the distal portion of the affected leg.
Generalized increases in venous pressure, with resultant
systemic edema, occur most commonly in congestive
heart failure affecting right ventricular cardiac function.
Although increased venous hydrostatic pressure is
contributory, the pathogenesis of cardiac edema is more
complex In congestive heart failure, reduced cardiac
output translates into reduced renal perfusion. Renal
hypoperfusion in turn triggers the renin-angiotensin-
aldosterone axis, inducing sodium and water retention by
the kidneys (secondary aldosteronism). This mechanism
normally functions to increase intravascular volume and
thereby improve cardiac output to restore normal renal
perfusion. However, if the failing heart cannot increase
cardiac output, the extra fluid load causes increased
venous pressure and, eventually, edema. Unless cardiac
output is restored or renal water retention reduced (e.g.,
by salt restriction, diuretics, or aldosterone antagonists), a
cycle of renal fluid retention and worsening edema
ensues. Although salt restriction, diuretics & aldosterone
antagonists are discussed here in the context of edema in
congestive heart failure, it should be understood that they
are also of value in the management of generalized edema
resulting from a variety of other causes.
Reduced Plasma Osmotic Pressure
Albumin is the serum protein most responsible for
maintaining intravascular colloid osmotic pressure; reduced
osmotic pressure occurs when albumin is inadequately
synthesized or is lost from the circulation. An important
cause of albumin loss is the nephrotic syndrome in which
glomerular capillary walls become leaky; patients typically
present with generalized edema. Reduced albumin synthesis
occurs in the setting of diffuse liver diseases (e.g., cirrhosis;
or due to protein malnutrition In each case, reduced plasma
osmotic pressure leads to a net movement of fluid into the
interstitial tissues with subsequent plasma volume
contraction. Predictably, reduced intravascular volume
leads to renal hypoperfusion followed by secondary
aldosteronism. Unfortunately, the retained salt and water
cannot correct the plasma volume deficit since the primary
defect of low serum proteins persists. As with congestive
heart failure, edema precipitated by low protein is
exacerbated by secondary salt and fluid retention.
Lymphatic Obstruction
Impaired lymphatic drainage and consequent lymphedema
is usually localized; it can result from inflammatory or
neoplastic obstruction. For example, the parasitic
infection filariasis can cause extensive inguinal lymphatic
and lymph node fibrosis. The resultant edema of the
external genitalia and lower limbs can be so massive as to

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
36
earn the appellation elephantiasis. Cancer of the breast
can be treated by resection and/or irradiation of the
associated axillary lymph nodes; the resultant scarring
and loss of lymphatic drainage can cause severe upper
extremity edema. In breast carcinoma infiltration and
obstruction of superficial lymphatics can also cause
edema of the overlying skin, the so-called peau d'orange
(orange peel) appearance. Such a finely pitted surface
results from an accentuation of depressions in the skin at
the site of hair follicles.
Sodium and Water Retention
Salt retention can also be a primary cause of edema.
Increased salt-with the obligate accompanying water-
causes both increased hydrostatic pressure (due to
expansion of the intravascular volume) and reduced
vascular osmotic pressure. Salt retention can occur with
any compromise of renal function, as in poststreptococcal
glomerulonephritis and acute renal failure
Morphology
Edema is most easily recognized grossly;
microscopically, edema fluid is reflected primarily as a
clearing and separation of the extracellular matrix
elements with subtle cell swelling. Although any organ or
tissue in the body may be involved, edema is most
commonly encountered in subcutaneous tissues, lungs,
and brain.
Subcutaneous edema can be diffuse or more prominent
in regions with high hydrostatic pressures; the ultimate
distribution depends on the underlying etiology. Even
diffuse edema is usually more prominent in certain body
areas as a result of the effects of gravity; a gravity-
dependent distribution is referred to as dependent edema
(e.g., involving the legs when standing, or involving the
sacrum when recumbent). Dependent edema is a
prominent feature of cardiac failure, particularly of
the right ventricle. Edema due to renal dysfunction or
nephrotic syndrome is generally more severe than
cardiac edema and affects all parts of the body equally.
Nevertheless, severe edema early in the disease course
can still manifest disproportionately in tissues with a
loose connective tissue matrix (e.g., the eyelids, causing
periorbital edema). Finger pressure over significantly
edematous subcutaneous tissue displaces the interstitial
fluid and leaves a finger-shaped depression, so-called
pitting edema.
Pulmonary edema is a common clinical problem most
frequently seen in the setting of left ventricular failure
(with a dependent distribution in the lungs), but it also
occurs in renal failure, acute respiratory distress
syndrome), pulmonary infections, and hypersensitivity
reactions. The lungs typically weigh two to three times
their normal weight, and sectioning reveals frothy,
sometimes blood-tinged fluid representing a mixture of
air, edema fluid, and extravasated red cells.
Edema of the brain may be localized to sites of focal
injury (e.g., infarct, abscesses or neoplasms) or may be
generalized, as in encephalitis, hypertensive crises, or
obstruction to the brain's venous outflow. Trauma may
result in local or generalized edema, depending on the
nature and extent of the injury. With generalized edema,
the brain is grossly swollen with narrowed sulci and
distended gyri showing signs of flattening against the
unyielding skull
Hyperemia and congestion
The terms hyperemia and congestion both indicate a local
increased volume of blood in a particular tissue. Hyperemia
is an active process resulting from augmented blood flow
due to arteriolar dilation (e.g., at sites of inflammation or in
skeletal muscle during exercise). The affected tissue is
redder than normal because of engorgement with
oxygenated blood. Congestion is a passive process resulting
from impaired venous return out of a tissue. It may occur
systemically, as in cardiac failure, or it may be local,
resulting from an isolated venous obstruction. The tissue
has a blue-red color (cyanosis), especially as worsening
congestion leads to accumulation of deoxygenated
hemoglobin in the affected tissues
Congestion of capillary beds is closely related to the
development of edema, so that congestion and edema
commonly occur together. In long-standing congestion,
called chronic passive congestion, the stasis of poorly
oxygenated blood causes chronic hypoxia, which in turn
can result in degeneration or death of parenchymal cells
and subsequent tissue fibrosis. Capillary rupture at such
sites of chronic congestion can also cause small foci of
hemorrhage; phagocytosis and catabolism of the
erythrocyte debris can result in accumulations of
hemosiderin-laden macrophages.
Morphology:-
Cut surfaces of hyperemic or congested tissues are
hemorrhagic and wet. Microscopically, acute pulmonary
congestion is characterized by alveolar capillaries
engorged with blood; there may also be associated
alveolar septal edema and/or focal minute intra-alveolar

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
37
hemorrhage. In chronic pulmonary congestion the septa
become thickened and fibrotic, and the alveolar spaces
may contain numerous hemosiderin-laden macrophages
("heart failure cells"). In acute hepatic congestion the
central vein and sinusoids are distended with blood, and
there may even be central hepatocyte degeneration; the
periportal hepatocytes, better oxygenated because of their
proximity to hepatic arterioles, undergo less severe
hypoxia and may develop only fatty change. In chronic
passive congestion of the liver the central regions of the
hepatic lobules are grossly red-brown and slightly
depressed (because of a loss of cells) and are accentuated
against the surrounding zones of uncongested tan,
sometimes fatty, liver ("nutmeg liver"; Microscopically,
there is centrilobular necrosis with hepatocyte drop-out,
hemorrhage, and hemosiderin-laden macrophages In long-
standing, severe hepatic congestion (most commonly
associated with heart failure), hepatic fibrosis ("cardiac
cirrhosis") can develop. It is important to note that
because the central portion of the hepatic lobule is the last
to receive blood, centrilobular necrosis can also occur
whenever there is reduced hepatic blood flow (including
shock from any cause); there need not be previous hepatic
congestion.
Hemorrhage
Hemorrhage is extravasation of blood from vessels into
the extravascular space. As described above, capillary
bleeding can occur under conditions of chronic
congestion; an increased tendency to hemorrhage (usually
with insignificant injury) occurs in a wide variety of
clinical disorders collectively called hemorrhagic
diatheses. Rupture of a large artery or vein results in
severe hemorrhage, and is almost always due to vascular
injury, including trauma, atherosclerosis, or inflammatory
or neoplastic erosion of the vessel wall.
Hemorrhage can be external or can be confined within a
tissue; any accumulation is referred to as a hematoma.
Hematomas can be relatively insignificant (e.g., a bruise)
or can involve so much bleeding as to cause death (e.g., a
massive retroperitoneal hematoma resulting from rupture
of a dissecting aortic aneurysm; Minute (1- to 2-mm)
hemorrhages into skin, mucous membranes, or serosal
surfaces are called petechiae) and are typically associated
with locally increased intravascular pressure, low platelet
counts (thrombocytopenia), defective platelet function, or
clotting factor deficiencies.Slightly larger (3- to 5-mm)
hemorrhages are called purpura and can be associated
with many of the same disorders that cause petechiae; in
addition, purpura can occur with trauma, vascular
inflammation (vasculitis), or increased vascular
fragility.Larger (1- to 2-cm) subcutaneous hematomas
(bruises) are called ecchymoses. The erythrocytes in these
local hemorrhages are phagocytosed and degraded by
macrophages; the hemoglobin (red-blue color) is
enzymatically converted into bilirubin (blue-green color)
and eventually into hemosiderin (golden-brown),
accounting for the characteristic color changes in a
hematoma.Large accumulations of blood in one or
another of the body cavities are called hemothorax,
hemopericardium, hemoperitoneum, or hemarthrosis (in
joints). Patients with extensive hemorrhages occasionally
develop jaundice from the massive breakdown of red
blood cells and systemic increases in bilirubin.
The clinical significance of hemorrhage depends on the
volume and rate of blood loss. Rapid removal of as much
as 20% of the blood volume or slow losses of even larger
amounts may have little impact in healthy adults; greater
losses, however, can cause hemorrhagic (hypovolemic)
shock (discussed later). The site of hemorrhage is also
important; bleeding that would be trivial in the
subcutaneous tissues may cause death if located in the
brain Finally, chronic or recurrent external blood loss
(e.g., a peptic ulcer or menstrual bleeding) causes a net
loss of iron, frequently culminating in an iron deficiency
anemia. In contrast, when red cells are retained (e.g., with
hemorrhage into body cavities or tissues), the iron can be
reutilized for hemoglobin synthesis.

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
38
Lecture 2+3 - Hemostasis &
thrombosis:-
Normal hemostasis is a consequence of tightly regulated
processes that maintain blood in a fluid, clot-free state in
normal vessels while inducing the rapid formation of a
localized hemostatic plug at the site of vascular injury.
The pathologic form of hemostasis is thrombosis; it
involves blood clot (thrombus) formation in uninjured
vessels or thrombotic occlusion of a vessel after relatively
minor injury. Both hemostasis and thrombosis involve
three components: the vascular wall, platelets, and the
coagulation cascade.
Normal Hemostasis
The sequence of events in hemostasis at a site of vascular
injury is shown in After initial injury a brief period of
arteriolar vasoconstriction occurs mostly as a result of
reflex neurogenic mechanisms and is augmented by the
local secretion of factors such as endothelin (a potent
endothelium-derived vasoconstrictor; The effect is
transient, and bleeding would resume were it not for
activation of the platelet and coagulation systems.
Endothelial injury also exposes highly thrombogenic
subendothelial extracellular matrix, allowing platelets to
adhere and be activated. Activation of platelets results in a
dramatic shape change (from small rounded disks to flat
plates with markedly increased surface area) & release of
secretory granules. Within minutes the secreted products
have recruited additional platelets (aggregation) to form a
hemostatic plug; this is the process of primary hemostasis
Tissue factor is also exposed at the site of injury. Also
known as factor III and thromboplastin, tissue factor is a
membrane-bound procoagulant glycoprotein synthesized
by endothelium. It acts in conjunction with factor VII , as
the major in vivo pathway to activate the coagulation
cascade, eventually culminating in thrombin generation.
Thrombin
cleaves circulating fibrinogen into insoluble
fibrin, creating a fibrin meshwork deposition. Thrombin
also induces further platelet recruitment& granule release.
This secondary hemostasis sequence lasts longer than the
initial platelet plug.
Polymerized fibrin and platelet aggregates form a solid
permanent plug to prevent any additional hemorrhage. At
this stage counter-regulatory mechanisms (e.g., tissue
plasminogen activator, t-PA) are set into motion to limit
the hemostatic plug to the site of injury
Endothelium
Endothelial cells modulate several (and frequently
opposing) aspects of normal hemostasis. The balance
between endothelial anti- and prothrombotic activities
determines whether thrombus formation, propagation, or
dissolution occurs. At baseline, endothelial cells exhibit
antiplatelet, anticoagulant, and fibrinolytic properties;
however, they are capable (after injury or activation) of
exhibiting numerous procoagulant activities It should also
be remembered that endothelium can be activated by
infectious agents, by hemodynamic factors, by plasma
mediators, and (most significantly) by cytokines
Antithrombotic Properties
Under most circumstances, endothelial cells maintain an
environment that promotes liquid blood flow by blocking
platelet adhesion and aggregation, by inhibiting the
coagulation cascade, and by lysing blood clots.
Antiplatelet Effects:-
An intact endothelium prevents platelets (and plasma
coagulation factors) from interacting with the highly
thrombogenic subendothelial ECM. Nonactivated platelets
do not adhere to the endothelium, a property intrinsic to the
plasma membrane of endothelium. Moreover, if platelets
are activated (e.g., after focal endothelial injury), they are
inhibited from adhering to the surrounding uninjured
endothelium by endothelial prostacyclin (PGI
2
) and nitric
oxide . Both mediators are potent vasodilators and
inhibitors of platelet aggregation; their synthesis by
endothelial cells is stimulated by several factors (e.g.,
thrombin and cytokines) produced during coagulation.
Endothelial cells also elaborate adenosine diphosphatase,
which degrades adenosine diphosphate (ADP) and further
inhibits platelet aggregation .
Anticoagulant Effects:-
Anticoagulant effects are mediated by membrane-
associated, heparin-like molecules and thrombomodulin
The heparin-like molecules act indirectly; they are
cofactors that allow antithrombin III to inactivate
thrombin , factor Xa, and several other coagulation
factors. Thrombomodulin also acts indirectly; it binds to
thrombin , converting it from a procoagulant to an
anticoagulant capable of activating the anticoagulant
protein C. Activated protein C, in turn, inhibits clotting by
proteolytic cleavage of factors Va and VIIIa; it requires
protein S, synthesized by endothelial cells, as a cofactor.
Fibrinolytic Properties:-
Endothelial cells synthesize tissue plasminogen activator
(t-PA), promoting fibrinolytic activity to clear fibrin
deposits from endothelial surfaces

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
39
Prothrombotic Properties:-
While endothelial cells exhibit properties that usually
limit blood clotting, they can also become prothrombotic,
with activities that affect platelets, coagulation proteins,
and the fibrinolytic system. Endothelial injury results in
platelet adhesion to subendothelial collagen; this occurs
through von Willebrand factor (vWF), an essential
cofactor for binding platelets to collagen and other
surfaces. vWF (both circulating and collagen bound) is
synthesized largely by normal endothelium. Loss of
endothelium exposes previously deposited vWF and
allows circulating vWF to also bind to the basement
membrane; in quick order, platelets adhere via their
glycoprotein Ib (GpIb) receptors
Cytokines such as tumor necrosis factor (TNF) or
interleukin-1 (IL-1) as well as bacterial endotoxin all
induce endothelial cell production of tissue factor, tissue
factor activates the extrinsic clotting pathway. By binding
activated IXa and Xa , endothelial cells augment the
catalytic activities of these coagulation factors. Finally,
endothelial cells also secrete plasminogen activator
inhibitors (PAIs), which depress fibrinolysis
Platelets:-
Platelets play a critical role in normal hemostasis. When
circulating and nonactivated they are membrane-bound
smooth disks expressing several glycoprotein receptors of
the integrin family and containing two types of granules:
α-Granules express the adhesion molecule P-selectin on
their membranes and contain fibrinogen, fibronectin,
factors V and VIII, platelet factor 4 (a heparin-binding
chemokine), platelet-derived growth factor (PDGF), and
transforming growth factor α (TGF-α).Dense bodies, or δ
granules, contain adenine nucleotides (ADP and ATP),
ionized calcium, histamine, serotonin, and epinephrine .
After vascular injury, platelets encounter ECM
constituents (of which collagen is the most important) and
additional proteins (vWF being critical) that are normally
not exposed when the endothelial layer is intact. Upon
contact with these proteins, platelets undergo three
reactions: (1) adhesion and shape change, (2) secretion
(release reaction), and (3) aggregation
Platelet Adhesion
Adhesion to ECM is mediated largely via interactions
with vWF acting as a bridge between platelet surface
receptors (e.g., GpIb) and exposed collagen Although
platelets can adhere directly to ECM, vWF-GpIb
associations are required to overcome the high shear
forces of flowing blood. Genetic deficiencies of vWF
(von Willebrand disease; or its receptors result in bleeding
disorders, highlighting the importance of these
interactions. Conversely, failure of the normal proteolytic
processing of vWF from high-molecular-weight
multimers to smaller forms leads to aberrant platelet
aggregation in the circulation; this defect in vWF
processing causes thrombotic thrombocytopenic purpura,
one of the so-called thrombotic microangiopathies
Secretion (Release Reaction)
Secretion of both granule types occurs soon after adhesion.
Various agonists can bind specific platelet surface receptors
and initiate an intracellular phosphorylation cascade that
leads to degranulation. Release of dense body contents is
especially important, since calcium is required in the
coagulation cascade and ADP is a potent mediator of
platelet aggregation (platelets adhering to other platelets-
discussed next). ADP also begets additional platelet ADP
release, amplifying the aggregation process. Finally,
platelet activation increases surface expression of
phospholipid complexes, which provide a critical nucleation
and binding site for calcium and coagulation factors in the
intrinsic clotting pathway
Platelet Aggregation
Aggregation follows platelet adhesion and granule
release. In addition to ADP, platelet-synthesized
thromboxane A
2
is also an important stimulus for platelet
aggregation. ADP and TXA
2
together drive an
autocatalytic process that promotes formation of an
enlarging platelet aggregate, the primary hemostatic plug.
This primary aggregation is reversible. However, with
activation of the coagulation cascade, the generation of
thrombin results in two processes that make an
irreversible hemostatic plug. Thrombin binds to a
platelet surface receptor (protease-activated receptor, or
PAR,; in association with ADP and TXA
2
, this interaction
induces further platelet aggregation. Platelet contraction
follows, creating an irreversibly fused mass of platelets
("viscous metamorphosis") constituting the definitive
secondary hemostatic plug. Concurrently, thrombin
converts fibrinogen to fibrin within and about the platelet
plug, contributing to the overall stability of the clot .
Both erythrocytes and leukocytes are also found in
hemostatic plugs; leukocytes adhere to platelets and
endothelium via adhesion molecules and contribute to the
inflammatory response that accompanies thrombosis.
Thrombin also contributes by directly stimulating neutrophil
and monocyte adhesion and by generating chemotactic fibrin
split products from the cleavage of fibrinogen
Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
40
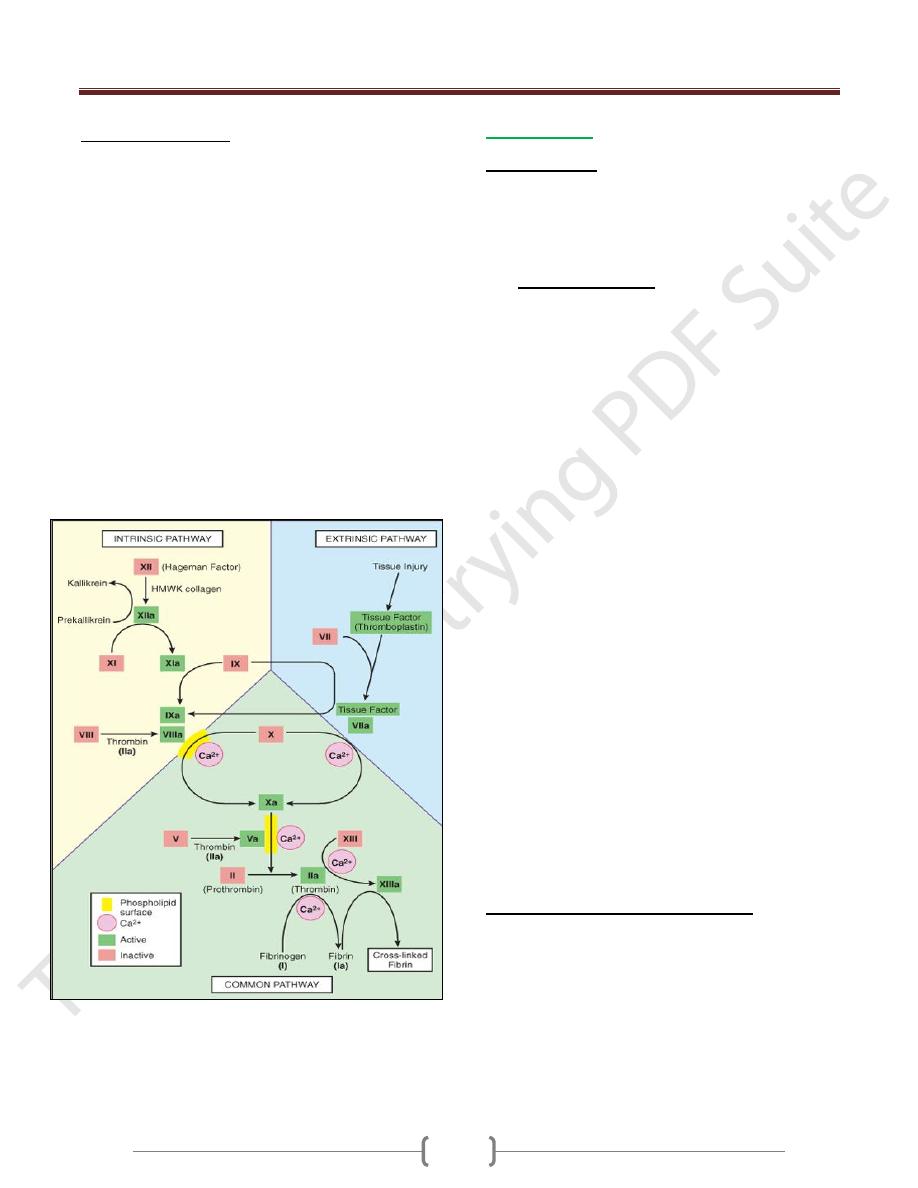
Coagulation Cascade
The coagulation cascade constitutes the third component
of the hemostatic process and is a major contributor to
thrombosis. The pathways are schematically presented in
below figure :-
Coagulation occurs via the sequential enzymatic
conversion of a cascade of circulating and locally
synthesized proteins. Tissue factor elaborated at sites of
injury is the most important initiator of the coagulation
cascade; at the final stage of coagulation, thrombin
converts fibrinogen into insoluble fibrin, which helps to
form the definitive hemostatic plug.Coagulation is
normally constrained to sites of vascular injury by:
Limiting enzymatic activation to phospholipid complexes
provided by activated plateletsNatural anticoagulants
elaborated at sites of endothelial injury or during
activation of the coagulation cascadeInduction of
fibrinolytic pathways involving plasmin through the
activities of various Pas.
Thrombosis
Pathogenesis:-
There are three primary influences on thrombus formation
(called Virchow's triad): (1) Endothelial injury (2) stasis
or turbulence of blood flow & (3) blood
hypercoagulability.
1- Endothelial Injury:
This is a dominant influence, since endothelial loss by itself
can lead to thrombosis. It is particularly important for
thrombus formation occurring in the heart or in the arterial
circulation, where the normally high flow rates might
otherwise hamper clotting by preventing platelet adhesion
or diluting coagulation factors. Thus, thrombus formation
within the cardiac chambers (e.g., after endocardial injury
due to myocardial infarction), over ulcerated plaques in
atherosclerotic arteries, or at sites of traumatic or
inflammatory vascular injury (vasculitis) is largely a
function of endothelial injury. Clearly, physical loss of
endothelium leads to exposure of subendothelial ECM,
adhesion of platelets, release of tissue factor, and local
depletion of PGI
2
and plasminogen activators. However, it
is important to note that endothelium need not be denuded
or physically disrupted to contribute to the development of
thrombosis; any perturbation in the dynamic balance of the
prothrombotic and antithrombotic activities of endothelium
can influence local clotting events Thus, dysfunctional
endothelium may elaborate greater amounts of
procoagulant factors (e.g., platelet adhesion molecules,
tissue factor, plasminogen activator inhibitors) or may
synthesize fewer anticoagulant effectors (e.g.,
thrombomodulin, PGI
2
, t-PA). Significant endothelial
dysfunction (in the absence of endothelial cell loss) may
occur with hypertension, turbulent flow over scarred valves,
or by the action of bacterial endotoxins. Even relatively
subtle influences, such as homocystinuria,
hypercholesterolemia, radiation, or products absorbed from
cigarette smoke, may be sources of endothelial dysfunction.
2 - Alterations in Normal Blood Flow:
Turbulence contributes to arterial and cardiac thrombosis
by causing endothelial injury or dysfunction, as well as by
forming countercurrents and local pockets of stasis; stasis
is a major contributor to the development of venous
thrombi. Normal blood flow is laminar, such that platelets
flow centrally in the vessel lumen, separated from the
endothelium by a slower moving clear zone of plasma.
Stasis and turbulence therefore:
Disrupt laminar flow and bring platelets into contact
with the endothelium

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
41
Prevent dilution of activated clotting factors by fresh-
flowing blood
Retard the inflow of clotting factor inhibitors and
permit the buildup of thrombi
Promote endothelial cell activation, resulting in local
thrombosis, leukocyte adhesion,
Turbulence and stasis contribute to thrombosis in several
clinical settings. Ulcerated atherosclerotic plaques not
only expose subendothelial ECM but also cause
turbulence. Abnormal aortic and arterial dilations, called
aneurysms, create local stasis and consequently a fertile
site for thrombosis Acute myocardial infarction results in
focally noncontractile myocardium; ventricular
remodeling after more remote infarction can lead to
aneurysm formation. In both cases cardiac mural thrombi
form more easily because of the local blood stasis Mitral
valve stenosis (e.g., after rheumatic heart disease) results
in left atrial dilation. In conjunction with atrial fibrillation,
a dilated atrium is a site of profound stasis and a prime
location for development of thrombi. Hyperviscosity
syndromes (such as polycythemia; ) increase resistance to
flow and cause small vessel stasis; the deformed red cells
in sickle cell anemia cause vascular occlusions, with the
resultant stasis also predisposing to thrombosis.
3- Hypercoagulability:
Hypercoagulability generally contributes less frequently
to thrombotic states but is nevertheless an important
component in the equation. It is loosely defined as any
alteration of the coagulation pathways that predisposes to
thrombosis, and it can be divided into primary (genetic)
and secondary (acquired) disorders.
Hypercoagulable States
Primary (Genetic)
Common
Mutation in factor V gene (factor V Leiden)
Mutation in prothrombin gene
Mutation in methyltetrahydrofolate gene
Rare
Antithrombin III deficiency
Protein C deficiency
Protein S deficiency
Very rare
Fibrinolysis defects
Secondary (Acquired)
High risk for thrombosis
Prolonged bed rest or immobilization
Myocardial infarction
Atrial fibrillation
Tissue damage (surgery, fracture, burns)
Cancer
Prosthetic cardiac valves
Disseminated intravascular coagulation
Heparin-induced thrombocytopenia
Antiphospholipid antibody syndrome (lupus
anticoagulant syndrome)
Lower risk for thrombosis
Cardiomyopathy
Nephrotic syndrome
Hyperestrogenic states (pregnancy)
Oral contraceptive use
Sickle cell anemia
Fate of the Thrombus:-
If a patient survives the initial thrombosis, in the ensuing
days or weeks thrombi undergo some combination of the
following four events:
1) Propagation. Thrombi accumulate additional platelets
and fibrin, eventually causing vessel obstruction.
2) Embolization. Thrombi dislodge or fragment and are
transported elsewhere in the vasculature.
3) Dissolution. Thrombi are removed by fibrinolytic activity.
4) Organization and recanalization. Thrombi induce
inflammation & fibrosis (organization). These can
eventually recanalize (re-establishing some degree of flow),
or they can be incorporated into a thickened vessel wall.
Morphology
Thrombi can develop anywhere in the cardiovascular
system (e.g., in cardiac chambers, on valves, or in
arteries, veins, or capillaries). The size and shape of a
thrombus depend on the site of origin and the cause.
Arterial or cardiac thrombi typically begin at sites of
endothelial injury or turbulence; venous thrombi
characteristically occur at sites of stasis. Thrombi are
focally attached to the underlying vascular surface;
arterial thrombi tend to grow in a retrograde direction
from the point of attachment, while venous thrombi
extend in the direction of blood flow (thus both tend to
propagate toward the heart). The propagating portion of a
thrombus tends to be poorly attached and therefore prone
to fragmentation, generating an embolus.

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
42
Thrombi can have grossly (and microscopically) apparent
laminations called lines of Zahn; these represent pale
platelet and fibrin layers alternating with darker
erythrocyte-rich layers. Such lines are significant only in
that they represent thrombosis in the setting of flowing
blood; their presence can therefore potentially distinguish
antemortem thrombosis from the bland nonlaminated
clots that occur in the postmortem state e. Although such
lines are typically not as apparent in veins or smaller
arteries (thrombi formed in sluggish venous flow usually
resemble statically coagulated blood), careful evaluation
generally reveals ill-defined laminations.
Thrombi occurring in heart chambers or in the aortic
lumen are designated mural thrombi. Abnormal
myocardial contraction (resulting from arrhythmias,
dilated cardiomyopathy, or myocardial infarction) or
endomyocardial injury (caused by myocarditis, catheter
trauma) promotes cardiac mural thrombi . while ulcerated
atherosclerotic plaques and aneurysmal dilation promote
aortic thrombosis.
Arterial thrombi are frequently occlusive and are
produced by platelet and coagulation activation; they are
typically a friable meshwork of platelets, fibrin,
erythrocytes, and degenerating leukocytes. Although
arterial thrombi are usually superimposed on an
atherosclerotic plaque, other vascular injury (vasculitis,
trauma) can be involved.
Venous thrombosis (phlebothrombosis) is almost
invariably occlusive, and the thrombus can create a long
cast of the lumen; venous thrombosis is largely the result
of activation of the coagulation cascade, and platelets play
a secondary role. Because these thrombi form in the
sluggish venous circulation, they also tend to contain
more enmeshed erythrocytes and are therefore called red,
or stasis, thrombi. The veins of the lower extremities are
most commonly affected (90% of venous thromboses);
however, venous thrombi can occur in the upper
extremities, periprostatic plexus, or ovarian and
periuterine veins; under special circumstances they may
be found in the dural sinuses, portal vein, or hepatic vein.
Postmortem clots can sometimes be mistaken at autopsy
for venous thrombi. However, postmortem "thrombi" are
gelatinous, with a dark red dependent portion where red
cells have settled by gravity, and a yellow "chicken fat"
supernatant, and they are usually not attached to the
underlying wall. In contrast, red thrombi are firmer and
are focally attached, and sectioning reveals strands of
gray fibrin.
Thrombi on heart valves are called vegetations. Bacterial
or fungal blood-borne infections can cause valve damage,
subsequently leading to large thrombotic masses
(infective endocarditis,). Sterile vegetations can also
develop on noninfected valves in hypercoagulable states,
so-called nonbacterial thrombotic endocarditis . Less
commonly, sterile, verrucous endocarditis (Libman-Sacks
endocarditis) can occur in the setting of systemic lupus
erythematosus

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
43
Lecture 4 - Embolism
An embolus is a detached intravascular solid, liquid, or
gaseous mass that is carried by the blood to a site distant
from its point of origin. Virtually 99% of all emboli
represent some part of a dislodged thrombus, hence the
term thromboembolism. Rare forms of emboli include fat
droplets, bubbles of air or nitrogen, atherosclerotic debris
(cholesterol emboli), tumor fragments, bits of bone
marrow, or foreign bodies such as bullets. However,
unless otherwise specified, an embolism should be
considered to be thrombotic in origin. Inevitably, emboli
lodge in vessels too small to permit further passage,
resulting in partial or complete vascular occlusion. The
consequences of thromboembolism include ischemic
necrosis (infarction) of downstream tissue. Depending on
the site of origin, emboli may lodge anywhere in the
vascular tree; the clinical outcomes are best understood
from the standpoint of whether emboli lodge in the
pulmonary or systemic circulations.
Pulmonary Thromboembolism
Pulmonary embolism has an incidence of 20 to 25 per
100,000 hospitalized patients. Although the rate of fatal
pulmonary emboli (as assessed at autopsy) has declined
from 6% to 2% over the last quarter century, pulmonary
embolism still causes about 200,000 deaths per year in the
United States. In more than 95% of cases, venous emboli
originate from deep leg vein thrombi above the level of
the knee . They are carried through progressively larger
channels and pass through the right side of the heart
before entering the pulmonary vasculature. Depending on
the size of the embolus, it may occlude the main
pulmonary artery, impact across the bifurcation (saddle
embolus), or pass out into the smaller, branching
arterioles . Frequently, there are multiple emboli, perhaps
sequentially, or as a shower of smaller emboli from a
single large thrombus; in general, the patient who has had
one pulmonary embolus is at high risk of having more.
Rarely, an embolus can pass through an interatrial or
interventricular defect, thereby entering the systemic
circulation (paradoxical embolism).
Most pulmonary emboli (60% to 80%) are clinically silent
because they are small. They eventually become
organized and become incorporated into the vascular
wall; in some cases, organization of the thromboembolus
leaves behind a delicate, bridging fibrous web.Sudden
death, right ventricular failure (cor pulmonale), or
cardiovascular collapse occurs when 60% or more of the
pulmonary circulation is obstructed with emboli. Embolic
obstruction of medium-sized arteries can cause pulmonary
hemorrhage but usually not pulmonary infarction because
the lung has a dual blood supply and the intact bronchial
arterial circulation continues to supply blood to the area.
However, a similar embolus in the setting of left-sided
cardiac failure (and resultant sluggish bronchial artery
blood flow) may result in a large infarct. Embolic
obstruction of small end-arteriolar pulmonary branches
usually does result in associated infarction. Many emboli
occurring over a period of time may cause pulmonary
hypertension with right ventricular failure
Systemic Thromboembolism
Systemic thromboembolism refers to emboli in the
arterial circulation. Most (80%) arise from intracardiac
mural thrombi, two-thirds of which are associated with
left ventricular wall infarcts and another quarter with
dilated left atria (e.g., secondary to mitral valve disease).
The remainder originate from aortic aneurysms, thrombi
on ulcerated atherosclerotic plaques, or fragmentation of
valvular vegetations . A very small fraction of systemic
emboli appear to arise in veins but end up in the arterial
circulation, through interventricular defects. These are
called paradoxical emboli. In contrast to venous emboli,
which tend to lodge primarily in one vascular bed (the
lung), arterial emboli can travel to a wide variety of sites;
the site of arrest depends on the point of origin of the
thromboembolus and the relative blood flow through the
downstream tissues. The major sites for arteriolar
embolization are the lower extremities (75%) and the
brain (10%), with the intestines, kidneys, and spleen
affected to a lesser extent. The consequences of
embolization in a tissue depend on vulnerability to
ischemia, caliber of the occluded vessel, and the collateral
blood supply; in general, arterial embolization causes
infarction of the affected tissues.
Fat Embolism
Microscopic fat globules can be found in the circulation
after fractures of long bones (which contain fatty marrow)
or after soft-tissue trauma. Fat enters the circulation by
rupture of the marrow vascular sinusoids or rupture of
venules in injured tissues. Although fat and marrow
embolism occurs in some 90% of individuals with severe
skeletal injuries, fewer than 10% of such patients show any
clinical findings. Fat embolism syndrome is characterized

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
44
by pulmonary insufficiency, neurologic symptoms, anemia,
and thrombocytopenia; it is fatal in about 10% of cases.
Typically, the symptoms appear 1 to 3 days after injury,
with sudden onset of tachypnea, dyspnea, and tachycardia.
Neurologic symptoms include irritability and restlessness,
with progression to delirium or coma.
The pathogenesis of fat emboli syndrome probably
involves both mechanical obstruction and biochemical
injury. Fat microemboli occlude pulmonary and cerebral
microvasculature; vascular occlusion is aggravated by
local platelet and erythrocyte aggregation. This pathology
is further exacerbated by free fatty acid release from the
fat globules, causing local toxic injury to endothelium.
Platelet activation and recruitment of granulocytes (with
free radical, protease, and eicosanoid release complete the
vascular assault. Because lipids are dissolved out of tissue
preparations by the solvents routinely used in paraffin
embedding, the microscopic demonstration of fat
microglobules (i.e., in the absence of accompanying
marrow) typically requires specialized techniques,
including frozen sections and fat stains.
Air Embolism
Gas bubbles within the circulation can obstruct vascular
flow (and cause distal ischemic injury) almost as readily as
thrombotic masses can. Air may enter the circulation during
obstetric procedures or as a consequence of chest wall
injury. Generally, more than 100 mL of air are required to
produce a clinical effect; bubbles can coalesce to form
frothy masses sufficiently large to occlude major vessels.
A particular form of gas embolism, called decompression
sickness, occurs when individuals are exposed to sudden
changes in atmospheric pressure. Scuba and deep-sea
divers, and underwater construction workers are at risk.
When air is breathed at high pressure (e.g., during a deep-
sea dive), increased amounts of gas (particularly nitrogen)
become dissolved in the blood and tissues. If the diver
then ascends (depressurizes) too rapidly, the nitrogen
expands in the tissues and bubbles out of solution in the
blood to form gas emboli that can induce focal ischemia
in a number of tissues, including brain and heart. The
rapid formation of gas bubbles within skeletal muscles
and supporting tissues in and about joints is responsible
for the painful condition called the bends (so named in the
1880s because afflicted individuals characteristically
arched their backs in a manner reminiscent of a then-
popular women's fashion called the Grecian Bend). In the
lungs, gas bubbles in the vasculature cause edema,
hemorrhages, and focal atelectasis or emphysema, leading
to respiratory distress, called the chokes. A more chronic
form of decompression sickness is called caisson disease,
where persistence of gas emboli in the bones leads to
multiple foci of ischemic necrosis; the heads of the
femurs, tibias, and humeri are most commonly affected.
Treating acute decompression sickness requires placing
the affected individual in a compression chamber to
increase barometric pressure and force the gas bubbles
back into solution. Subsequent slow decompression
theoretically permits gradual resorption and exhalation of
the gases so that obstructive bubbles do not re-form.
Amniotic Fluid Embolism:-
Amniotic fluid embolism is a grave but fortunately
uncommon complication of labor and the immediate
postpartum period (1 in 50,000 deliveries). It has a
mortality rate in excess of 20% to 40%. The onset is
characterized by sudden severe dyspnea, cyanosis, and
hypotensive shock, followed by seizures and coma. If the
patient survives the initial crisis, pulmonary edema
typically develops, along with (in half the patients)
disseminated intravascular coagulation (DIC), due to
release of thrombogenic substances from amniotic fluid.
The underlying cause is entry of amniotic fluid (and its
contents) into the maternal circulation via a tear in the
placental membranes and rupture of uterine veins.
Classically, there is marked pulmonary edema and diffuse
alveolar damage with the pulmonary microcirculation
containing squamous cells shed from fetal skin, lanugo
hair, fat from vernix caseosa, and mucin derived from the
fetal respiratory or gastrointestinal tracts. Systemic fibrin
thrombi indicate the onset of DIC.

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
45
Lecture 5 – Infarction & Shock
Infarction
An infarct is an area of ischemic necrosis caused by
occlusion of either the arterial supply or the venous
drainage in a particular tissue. Tissue infarction is a
common and extremely important cause of clinical illness.
More than half of all deaths in the United States are
caused by cardiovascular disease, and most of these are
attributable to myocardial or cerebral infarction.
Pulmonary infarction is a common complication in
several clinical settings, bowel infarction is frequently
fatal, and ischemic necrosis of the extremities (gangrene)
is a serious problem in the diabetic population.
Nearly 99% of all infarcts result from thrombotic or
embolic events, and almost all result from arterial
occlusion. Occasionally, infarction may also be caused by
other mechanisms, such as local vasospasm, expansion of
an atheroma secondary to intraplaque hemorrhage, or
extrinsic compression of a vessel (e.g., by tumor).
Uncommon causes include vessel twisting (e.g., in
testicular torsion or bowel volvulus), vascular compression
by edema or entrapment in a hernia sac, or traumatic vessel
rupture. Although venous thrombosis can cause infarction,
it more often merely induces venous obstruction and
congestion. Usually, bypass channels open rapidly after the
occlusion forms, providing some outflow from the area
that, in turn, improves the arterial inflow. Infarcts caused by
venous thrombosis are more likely in organs with a single
venous outflow channel (e.g., testis and ovary).
Morphology
Infarcts are classified on the basis of their color (reflecting
the amount of hemorrhage) & the presence or absence of
microbial infection. Therefore, infarcts may be either red
(hemorrhagic) or white (anemic) & may be either septic or
bland.
Red infarcts
occur (1) with venous occlusions (such as in
ovarian torsion); (2) in loose tissues (such as lung) that
allow blood to collect in the infarcted zone; (3) in tissues
with dual circulations such as lung and small intestine,
permitting flow of blood from an unobstructed parallel
supply into a necrotic area (such perfusion not being
sufficient to rescue the ischemic tissues); (4) in tissues that
were previously congested because of sluggish venous
outflow; (5) when flow is re-established to a site of
previous arterial occlusion & necrosis (e.g., fragmentation
of an occlusive embolus or angioplasty of a thrombotic
lesion).
White infarcts
occur with arterial occlusions or in solid
organs (such as heart, spleen, and kidney), where the
solidity of the tissue limits the amount of hemorrhage that
can seep into the area of ischemic necrosis from adjoining
capillary beds.
All infarcts tend to be wedge shaped, with the occluded
vessel at the apex and the periphery of the organ forming
the base, when the base is a serosal surface there can be
an overlying fibrinous exudate. At the outset, all infarcts
are poorly defined and slightly hemorrhagic. The margins
of both types of infarcts tend to become better defined
with time by a narrow rim of congestion attributable to
inflammation at the edge of the lesion.
Factors that influence development of an infarct
Vascular occlusion can have no or minimal effect, or can
cause death of a tissue or even the individual. The major
determinants of the eventual outcome include the nature
of the vascular supply, the rate of development of the
occlusion, vulnerability to hypoxia, and the oxygen
content of blood
Nature of the Vascular Supply
The availability of an alternative blood supply is the most
important determinant of whether occlusion of a vessel will
cause damage. For example, as mentioned above, lungs
have a dual pulmonary and bronchial artery blood supply;
thus, obstruction of small pulmonary arterioles does not
cause infarction in an otherwise healthy individual with an
intact bronchial circulation. Similarly, the liver, with its
dual hepatic artery and portal vein circulation, and the hand
and forearm, with their dual radial and ulnar arterial supply,
are all relatively resistant to infarction. In contrast, renal
and splenic circulations are end-arterial, and obstruction of
such vessels generally causes infarction.
Rate of Development of Occlusion
Slowly developing occlusions are less likely to cause
infarction because they provide time for the development
of alternative perfusion pathways. For example, small
interarteriolar anastomoses-normally with minimal
functional flow-interconnect the three major coronary
arteries in the heart. If one of the coronaries is slowly
occluded (e.g., by an encroaching atherosclerotic plaque),
flow within this collateral circulation may increase
sufficiently to prevent infarction, even though the major
coronary artery is eventually occluded.
Vulnerability to Hypoxia
The susceptibility of a tissue to hypoxia influences the
likelihood of infarction. Neurons undergo irreversible
damage when deprived of their blood supply for only 3 to

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
46
4 minutes. Myocardial cells, though hardier than neurons,
are also quite sensitive and die after only 20 to 30 minutes
of ischemia. In contrast, fibroblasts within myocardium
remain viable after many hours of ischemia.
Oxygen Content of Blood
The partial pressure of oxygen in blood also determines
the outcome of vascular occlusion. Partial flow
obstruction of a small vessel in an anemic or cyanotic
patient might lead to tissue infarction, whereas it would be
without effect under conditions of normal oxygen tension.
In this way congestive heart failure, with compromised
flow and ventilation, could cause infarction in the setting
of an otherwise inconsequential blockage.
Shock
Shock is the final common pathway for a number of
potentially lethal clinical events, including severe
hemorrhage, extensive trauma or burns, large myocardial
infarction, massive pulmonary embolism, and microbial
sepsis. Regardless of the underlying pathology, shock
gives rise to systemic hypoperfusion; it can be caused
either by reduced cardiac output or by reduced effective
circulating blood volume. The end results are
hypotension, impaired tissue perfusion, and cellular
hypoxia. Although the hypoxic and metabolic effects of
hypoperfusion initially cause only reversible cellular
injury, persistence of shock eventually causes irreversible
tissue injury and can culminate in the death of the patient.
There are three general categories of shock
:
cardiogenic, hypovolemic, and septic (Table 4-3). The
mechanisms underlying cardiogenic and hypovolemic
shock are fairly straightforward; septic shock is
substantially more complicated and is discussed in further
detail below.
1) Cardiogenic shock results from failure of the cardiac
pump. This may be caused by myocardial damage
(infarction), ventricular arrhythmias, extrinsic
compression (cardiac tamponade, Chapter 11), or outflow
obstruction (e.g., pulmonary embolism).
2) Hypovolemic shock results from loss of blood or plasma
volume. This may be caused by hemorrhage, fluid loss
from severe burns, or trauma.
3) Septic shock is caused by microbial infection. Most
commonly this occurs in the setting of gram-negative
infections (endotoxic shock), but it can also occur with
gram-positive and fungal infections. Notably, there need
not be systemic bacteremia to induce septic shock; host
inflammatory responses to local extravascular infections
may be sufficient (see below).
Less commonly, shock may occur in the setting of an
anesthetic accident or a spinal cord injury (neurogenic
shock), as a result of loss of vascular tone and peripheral
pooling of blood. Anaphylactic shock represents systemic
vasodilation and increased vascular permeability caused by
an immunoglobulin E hypersensitivity reaction (Chapter 5).
In these situations, acute severe widespread vasodilation
results in tissue hypoperfusion & cellular anoxia.
Pathogenesis of Septic Shock
With a 25% to 50% mortality rate, septic shock ranks first
among the causes of death in intensive care units and
accounts for more than 200,000 deaths annually in the
United States. Moreover, the continuing increase in the
incidence of sepsis syndromes is attributable to improved
life support for high-risk patients, an increase in invasive
procedures, and the growing numbers of
immunocompromised hosts (secondary to chemotherapy,
immunosuppression, or infection with the human
immunodeficiency virus). Septic shock results from the
host innate immune response to infectious organisms that
may be blood borne or localized to a particular site.
Most cases of septic shock (approximately 70%) are
caused by endotoxin-producing gram-negative bacilli
(Chapter 9)-hence the term endotoxic shock. Endotoxins
are bacterial wall lipopolysaccharides (LPS) consisting of
a toxic fatty acid (lipid A) core common to all gram-
negative bacteria, and a complex polysaccharide coat
(including O antigen) unique for each species. Analogous
molecules in the walls of gram-positive bacteria and fungi
can also elicit septic shock.
All of the cellular and hemodynamic effects of septic shock
can be reproduced by LPS injection alone. Free LPS
attaches to a circulating LPS-binding protein, and the
complex then binds to a specific receptor (CD14) on
monocytes, macrophages, and neutrophils. Engagement of
CD14 (even at doses as minute as 10 pg/mL) results in
intracellular signaling via an associated "Toll-like receptor"
protein 4 (TLR-4), resulting in profound activation of
mononuclear cells and production of potent effector
cytokines such as IL-1 and TNF (Chapter 2). These
cytokines act on endothelial cells and have a variety of
effects including reduced synthesis of anticoagulation
factors such as tissue factor pathway inhibitor & thrombo-
modulin (see Fig. 4-7). The effects of the cytokines may be
amplified by TLR-4 engagement on endothelial cells.

Unit 3: Hemodynamic Disorders, Thromboembolism, and Shock
47
TLR-mediated activation helps to trigger the innate
immune system to efficiently eradicate invading microbes
(Chapter 5). Unfortunately, depending on the dosage and
the extent of immune and vascular activation, the
secondary effects of LPS release can also cause severe
pathologic changes, including fatal shock.
At low doses, LPS predominantly activates monocytes,
macrophages, and neutrophils; it can also directly activate
complement, thereby contributing to local eradication of
bacteria. Mononuclear phagocytes respond to LPS by
producing TNF, which in turn induces IL-1 synthesis.
Both TNF and IL-1 act on endothelial cells (and other cell
types) to produce additional cytokines (e.g., IL-6 and IL-
8) and induce adhesion molecules (Chapter 2). Thus, the
initial release of LPS results in a circumscribed cytokine
cascade that enhances the local acute inflammatory
response and improves clearance of the infection.
With moderately severe infections, and therefore with
higher levels of LPS (and a consequent augmentation of
the cytokine cascade), cytokine-induced secondary
effectors (e.g., nitric oxide and platelet-activating factor;
Chapter 2) become significant. In addition, systemic
effects of TNF and IL-1 may begin to be seen, including
fever, increased synthesis of acute-phase reactants, and
increased production of circulating neutrophils (see Fig.
4-21). Higher LPS levels tip the endothelium toward a net
procoagulant phenotype.
Finally, at still higher levels of LPS, the syndrome of
septic shock supervenes (see Fig. 4-21); the same
cytokine and secondary mediators, now at high levels,
result in Systemic vasodilation (hypotension)Diminished
myocardial contractilityWidespread endothelial injury and
activation, causing systemic leukocyte adhesion and
diffuse alveolar capillary damage in the lung (Chapter
13)Activation of the coagulation system, culminating in
disseminated intravascular coagulation (DIC)
The hypoperfusion resulting from the combined effects of
widespread vasodilation, myocardial pump failure, and
DIC causes multiorgan system failure that affects the
liver, kidneys, and central nervous system, among others.
Unless the underlying infection (and LPS overload) is
rapidly brought under control, the patient usually dies. In
some experimental animal models, soluble CD14,
antibodies to LPS-binding proteins, or pharmacologic
inhibitors of the secondary mediators (e.g., nitric oxide
synthesis) have demonstrated some efficacy in protecting
against septic shock. Unfortunately, these interventions
have not yet proved of significant clinical benefit in
patients, perhaps because many different pathways and
mediators are activated by LPS.
An interesting group of bacterial proteins called
superantigens also causes a syndrome similar to septic
shock (e.g., toxic shock syndrome toxin 1, responsible for
the toxic shock syndrome). Superantigens are polyclonal
T-lymphocyte activators that induce systemic
inflammatory cytokine cascades similar to those that
occur in response to LPS. Their actions can result in a
variety of clinical manifestations ranging from a diffuse
rash to vasodilation, hypotension, and death.
Stages of Shock
An initial nonprogressive stage during which reflex
compensatory mechanisms are activated and perfusion of
vital organs is maintained
A progressive stage characterized by tissue hypoperfusion
and onset of worsening circulatory and metabolic
imbalances
An irreversible stage that sets in after the body has incurred
cellular and tissue injury so severe that even if the
hemodynamic defects are corrected, survival is not possible
Morphology
The cellular and tissue changes induced by shock are
essentially those of hypoxic injury (Chapter 1), due to some
combination of hypoperfusion and microvascular thrombosis.
Since shock is characterized by failure of many organ
systems, the cellular changes may appear in any tissue.
Nevertheless, they are particularly evident in the brain, heart,
kidneys, adrenal glands, and gastrointestinal tract. Fibrin
thrombi may be identified in virtually any tissue, although
they are usually most readily visualized in kidney glomeruli.
The adrenal changes in shock are those seen in all forms of
stress; essentially there is cortical cell lipid depletion. This
reflects not adrenal exhaustion but instead conversion of the
relatively inactive vacuolated cells to metabolically active
cells that use stored lipids for the synthesis of steroids. The
kidneys typically reveal acute tubular necrosis (Chapter 14)
so that oliguria, anuria, and electrolyte disturbances dominate
the clinical picture. The gastrointestinal tract may mainfest
focal mucosal hemorrhage and necrosis. The lungs are
seldom affected in pure hypovolemic shock, because they are
somewhat resistant to hypoxic injury. However, when shock
is caused by bacterial sepsis or trauma, changes of diffuse
alveolar damage may develop, the so-called shock lung.
With the exception of neuronal and myocyte ischemic loss,
virtually all tissues may revert to normal if the patient
survives. Unfortunately, most patients with irreversible
changes due to severe shock die before the tissues can
recover.