
1
Interstitial and Infiltrative Pulmonary Diseases
Diffuse parenchymal lung disease
The diffuse parenchymal lung diseases (DPLDs) are a heterogeneous group of conditions
associated with diffuse thickening of the alveolar walls with:
1. Inflammatory cells and exudates (e.g. the acute respiratory distress syndrome-
ARDS),
2. Granulomas (e.g. sarcoidosis),
3. Alveolar haemorrhage (e.g. Goodpasture's syndrome,)
4. And/or fibrosis (e.g. fibrosing alveolitis).
Diagnosis of interstitial lung disease:
1-A general approach:
Establishing a diagnosis is important because:
•
Firstly
, there are prognostic implications; for example, sarcoidosis is frequently
self-limiting, whereas idiopathic pulmonary fibrosis (IPF) is most often fatal.
•
Secondly
, establishing a specific diagnosis will avoid inappropriate treatment; for
example, the powerful immunosuppressive regimens used for some cases of IPF
would be undesirable if the underlying condition was asbestosis or hypersensitivity
pneumonitis.
•
Thirdly
, some DPLDs can be expected to respond better than others to
treatment, e.g. a good symptomatic response to corticosteroids could be predicted
in sarcoidosis, whereas the prognosis would need to be more guarded in IPF.
•
Finally
, a lung biopsy taken when the patient is already established on empirical
immunosuppressive therapy is not only associated with a higher morbidity and
mortality, but the interpretation of the tissue obtained is more difficult.
•
It is desirable, therefore, to be confident about the diagnosis before starting any
therapy.
The smoking status should be recorded and a drug history that includes over-the-
counter prescriptions should be obtained.

2
A history of rashes
, joint pains or renal disease may suggest an underlying connective
tissue disorder or vasculities.
The presence of any comorbid disease should be ascertained such as collagen vascular
disease, immunodeficiency, HIV or malignancy.
In exceptional cases there is a family history of DPLD.
2-Physical signs
In many cases, especially in early disease, there are few, if any, physical signs.
In
advanced disease, tachypnoea and cyanosis may be evident at rest and there may be
signs of pulmonary hypertension and right heart failure.
Finger clubbing may be prominent,
particularly in IPF or asbestosis
. There may be
restriction of lung expansion and showers of end-inspiratory crackles posteriorly and
laterally.
Extrapulmonary signs, including lymphadenopathy or uveitis, may be present in
sarcoidosis and
arthropathies or rashes may occur when a DPLD is a manifestation of a
connective tissue disorder.
Conditions Which Mimic Interstitial Lung Diseases
1-Infection
•
Viral pneumonia
•
Pneumocystis jirovecii
•
Mycoplasma pneumoniae
•
TB
•
Parasites, e.g. filariasis
•
Fungal infection
2-Malignancy
•
Leukaemia and lymphoma
•
Lymphatic carcinomatosis
•
Multiple metastases
•
Bronchoalveolar carcinoma

3
3-Pulmonary oedema
4-Aspiration pneumonitis
Investigations
Laboratory investigations:
•
Full blood count-lymphopenia in sarcoid; eosinophilia in pulmonary eosinophilias and
drug reactions; neutrophilia in hypersensitivity pneumonitis
•
Ca2-+may be elevated in sarcoid
•
Lactate dehydrogenase (LDH)-may provide non-specific indicator of disease
activity in DPLD
•
Serum ACE-non-specific indicator of disease activity in sarcoid
•
ESR and CRP may be non-specifically raised
•
Autoimmune screen and rheumatoid factor may suggest collagen vascular disease
Radiology:
Chest X-ray,High-resolution CT scan
Pulmonary function:
Spirometry, lung volumes, gas transfer, exercise tests Pulmonary
function tests typically show a restrictive pattern with diminished lung volumes and a
reduced gas transfer, although an elevated gas transfer may be seen in cases of alveolar
haemorrhage.
Bronchoscopy
•
Bronchoalveolar lavage-infection, differential cell counts
•
Bronchial biopsy may be useful in sarcoid
Lung biopsy (in selected cases)
•
Transbronchial biopsy useful in sarcoid and differential of malignancy or infection
•
Video-assisted thoracoscopy (VATS)
Others
•
Liver biopsy
•
Urinary calcium excretion may be useful in sarcoid

4
Idiopathic interstitial pneumonias
The idiopathic interstitial pneumonias (IIPs) are
characterised by varying patterns of
inflammation and fibrosis in the lung parenchyma
, and comprise a number of
clinicopathological entities that are sufficiently different from one another to be
considered as separate diseases.
The most common and important of these is idiopathic pulmonary fibrosis (IPF),
which
accounts for over 85% of new cases.
Idiopathic pulmonary fibrosis
•
Idiopathic pulmonary fibrosis refers to a
specific form of DPLD characterised by
pathological (or HRCT) evidence of 'usual interstitial pneumonia' (UIP).
•
The histological features are suggestive of repeated episodes of focal damage to the
alveolar epithelium consistent with an autoimmune process, but the
aetiology
remains
elusive; putative factors include viruses (e.g. Epstein-Barr virus), occupational dusts
(metal or wood), drugs (antidepressants) or chronic gastro-oesophageal reflux.
•
Familial cases are rare but genetic factors that control the inflammatory and fibrotic
response are likely to be important.
•
There is a strong association with cigarette smoking.
Clinical features
•
IPF is uncommon before the age of 50 years. It usually presents with
insidiously
progressive breathlessness and a non-productive cough.
•
Constitutional symptoms are unusual. Clinical findings include
finger clubbing and the
presence of bi-basal fine late inspiratory crackles.
•
The natural history is usually one of steady decline, but some patients are prone to
exacerbations which are accompanied by an acute deterioration in breathlessness,
disturbed gas exchange, and new
ground glass changes or consolidation on HRCT.
•
In advanced disease
, central cyanosis is detectable and patients may develop features
of right heart failure.
•
There is no circulating marker for IPF.

5
•
Non-specific findings include hypergammaglobulinaemia, positive rheumatoid factor or
antinuclear factor, and an elevated LDH which may reflect active pneumonitis.
Investigations:
•
Pulmonary function tests classically show a restrictive defect with reduced lung
volumes and gas transfer.
•
However, lung volumes may be paradoxically preserved in patients with concomitant
emphysema.
•
Dynamic tests are useful to document exercise tolerance and demonstrate exercise-
induced arterial hypoxaemia, but as IPF advances, arterial hypoxaemia and hypocapnia
are present at rest.
•
Virtually all patients have an abnormal chest X-ray at presentation with lower zone bi-
basal reticular and reticulonodular opacities.
•
A 'honeycomb' appearance may be seen in advanced disease but is non-specific.
•
HRCT typically demonstrates a patchy, predominantly peripheral, subpleural and basal
reticular pattern with subpleural cysts (honeycombing) and/or traction bronchiectasis.
•
A lung biopsy is not usually required in those with typical clinical features and HRCT
appearances, particularly if other known causes of interstitial lung disease have been
excluded, but should be considered in cases of diagnostic uncertainty or with atypical
features.
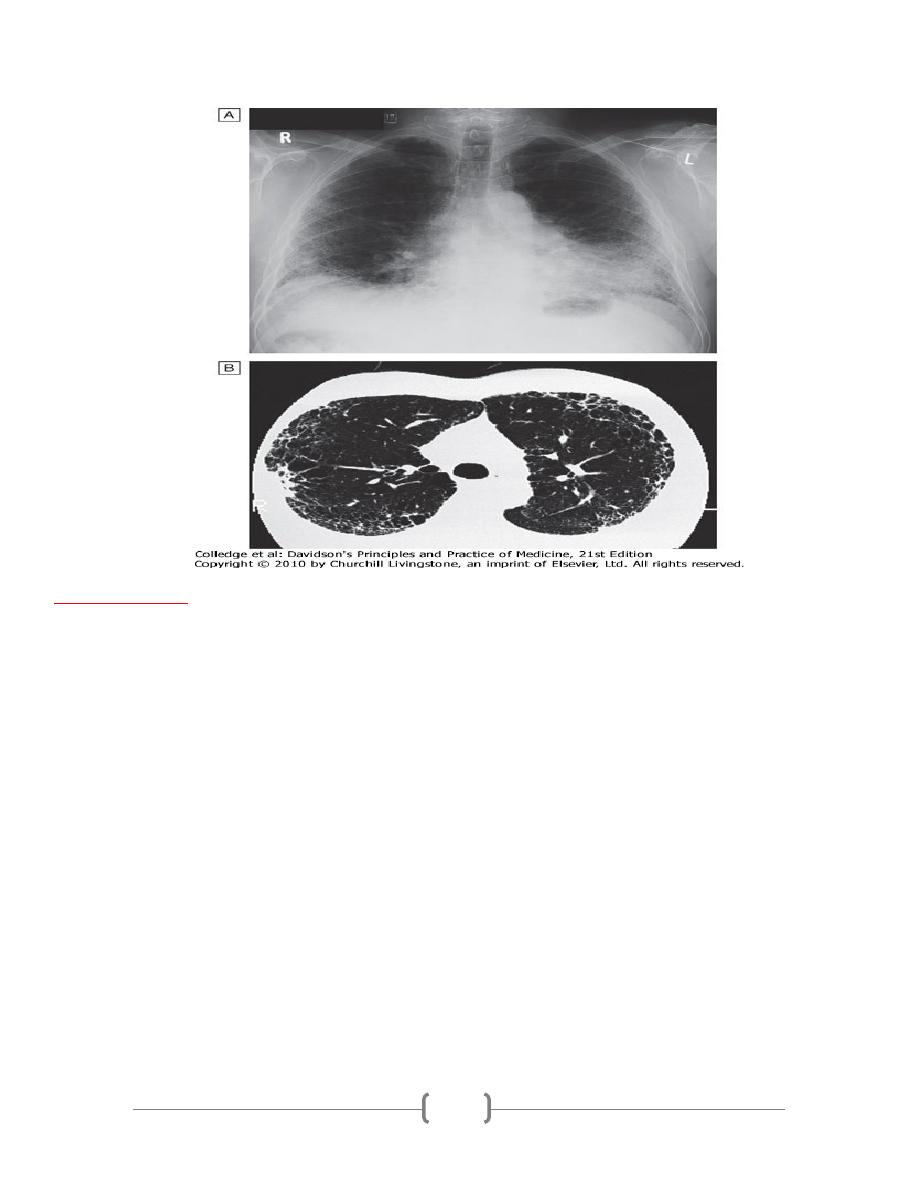
6
Management
Treatment is challenging. Prednisolone therapy (0.5 mg/kg) combined with azathioprine
(2-3 mg/kg) is advocated for patients who are:
1. highly symptomatic or
2. have rapidly progressive disease,
3. have a predominantly 'ground glass' appearance on CT or
4. a sustained fall of > 15% in their FVC or gas transfer over a 3-6-month period.
•
However, response rates are notoriously poor, side-effects are guaranteed.
•
If objective evidence of improvement can be demonstrated, the prednisolone
dose may be gradually reduced to a maintenance dose of 10-12.5 mg daily.
•
The potential of therapies such as warfarin, N-acetylcysteine, IFN-γ1b, bosentan
is being explored but cannot be recommended outside clinical trials at present.
•
Lung transplantation should be considered in young patients with advanced disease.
•
Oxygen may be provided for palliation of breathlessness but opiates may be
required for relief of severe dyspnoea.
•
The optimum treatment for acute exacerbations is unknown.

7
Prognosis
•
A median survival of 3 years is widely quoted; however, the rate of disease
progression varies considerably from death within a few months to survival with
minimal symptoms for many years.
•
Serial lung function testing may provide useful prognostic information, with
relative preservation of lung function
suggesting longer survival and desaturation
on exercise heralding a poorer prognosis.
•
The finding of high numbers of fibroblastic foci on biopsy suggests a more rapid
deterioration.
•
IPF is associated with
an increased risk of carcinoma of the lung.
Sarcoidosis
•
Sarcoidosis
is a multisystem granulomatous disorder of unknown aetiology that is
characterised by the presence of non-caseating granulomas.
•
The condition is more frequently described in colder parts of northern Europe.
•
It also appears to be more common and more severe in those from a West Indian or
Asian background; Eskimos, Arabs and Chinese are rarely affected.
•
The tendency for sarcoid to present in the spring and summer has led to speculation
as to the role of infective agents, including mycobacteria, propionibacteria and
viruses, but the cause remains elusive.
•
Genetic susceptibility
is supported by familial clustering; a range of
class II HLA
alleles confer protection from or susceptibility to the condition.
•
Sarcoidosis occurs less frequently in
smokers.
Clinical features
•
Sarcoidosis is considered with other DPLD as over 90% of cases affect the lungs, but
the condition can affect almost any organ.
•
Löfgren's syndrome
-an acute illness characterised by erythema nodosum, peripheral
arthropathy, uveitis, bilateral hilar lymphadenopathy (BHL), lethargy and occasionally
fever-is often seen in young adults.

8
•
Alternatively, BHL may be detected in an otherwise asymptomatic individual
undergoing a chest X-ray for other purposes.
•
Pulmonary disease may also present in a more insidious manner with cough, exertional
breathlessness and radiographic infiltrates; chest auscultation is often surprisingly
unremarkable.
•
Fibrosis occurs in around
20% of cases
of pulmonary sarcoidosis and may cause
a silent
loss of lung function.
•
Pleural disease is uncommon and finger clubbing is not a feature
.
•
Complications such as bronchiectasis, aspergilloma, pneumothorax, pulmonary
hypertension and cor pulmonale have been reported but are rare.
Presentation of sarcoidosis
•
Asymptomatic: abnormal routine chest X-ray ( 30%) or abnormal liver function tests
•
Respiratory and constitutional symptoms (20-30%)
•
Erythema nodosum and arthralgia (20-30%)
•
Ocular symptoms (5-10%)
•
Skin sarcoid (including lupus pernio) (5%)
•
Superficial lymphadenopathy (5%)
•
Other (1%), e.g. hypercalcaemia, diabetes insipidus, cranial nerve palsies, cardiac
arrhythmias, nephrocalcinosis
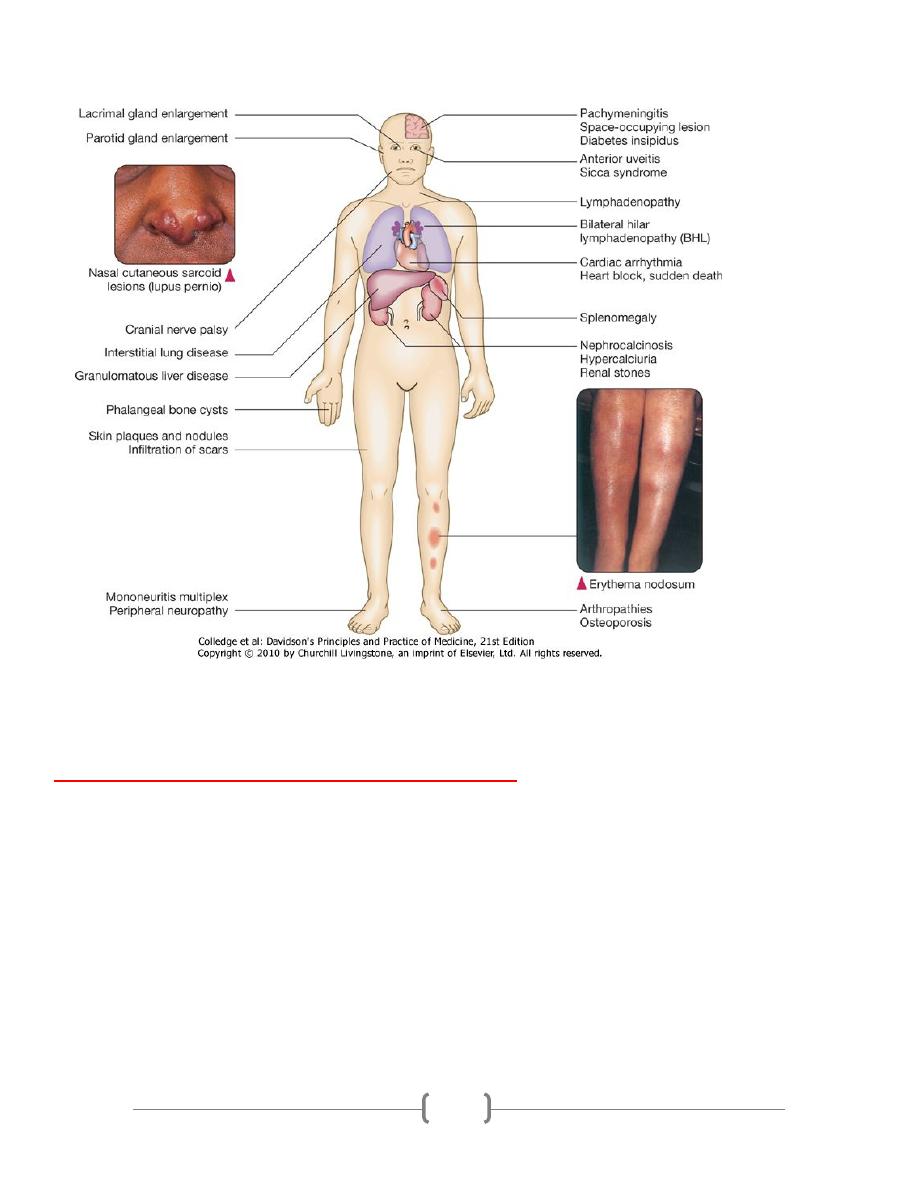
9
Chest X-ray changes in sarcoidosis
Stage I:
BHL (usually symmetrical); paratracheal nodes often enlarged
Often asymptomatic, but may be associated with erythema nodosum and
arthralgia. The majority of cases resolve spontaneously within 1 year
Stage II:
BHL and parenchymal infiltrates
Patients may present with breathlessness or
cough. The majority of cases resolve spontaneously
Stage III:
parenchymal infiltrates without BHL
Disease less likely to resolve spontaneously
Stage IV:

10
pulmonary fibrosis
Can cause progression to ventilatory failure, pulmonary
hypertension and cor pulmonale
Investigations
•
Lymphopenia is characteristic and liver function tests may be mildly deranged
.
Hypercalcaemia may be present (reflecting increased formation of calcitriol-1,25-
dihydroxyvitamin D
3
-by alveolar macrophages), particularly if the patient has been
exposed to strong sunlight.
•
Hypercalciuria
may also be seen and may lead to nephrocalcinosis.
•
Serum angiotensin-converting enzyme
(ACE)
is a non-specific marker of disease
activity and can assist in monitoring the clinical course.
•
Chest radiography
has been used to stage sarcoid.
•
In patients with pulmonary infiltrates,
pulmonary function testing
may show a
restrictive defect accompanied by
impaired gas exchange
.
•
Exercise tests may reveal oxygen desaturation.
•
Transbronchial (and bronchial) biopsies
show non-caseating granulomas
and the
mucosa may have a 'cobblestone' appearance at bronchoscopy.
•
The BAL fluid typically contains an increased CD4:CD8 T-cell ratio.
•
Characteristic HRCT appearances include reticulonodular opacities.
•
The occurrence of erythema nodosum with BHL on chest X-ray is often
sufficient for a confident diagnosis, without recourse to a tissue biopsy.
•
Similarly, a typical presentation with classical HRCT features may also be
accepted.
•
However, in other instances the diagnosis should be confirmed by histological
examination of the involved organ.
•
The presence of anergy (e.g. to tuberculin skin tests) may support the diagnosis.

11
Management
•
Patients who present with acute illness and erythema nodosum should receive
NSAIDs and, if symptoms are severe, a short course of corticosteroids.
•
The majority of patients enjoy
spontaneous remission
and so if there is no
evidence of organ damage, systemic corticosteroid therapy can be withheld for 6
months.
•
However, prednisolone
(at a starting dose of 20-40 mg/day) should be commenced
immediately in the presence of hypercalcaemia, pulmonary impairment, renal
impairment and uveitis
.
•
Topical steroids may be useful in cases of mild uveitis, and inhaled corticosteroids
have been used to shorten the duration of systemic corticosteroid use in
asymptomatic parenchymal sarcoid.
•
Patients should be warned that strong sunlight may precipitate hypercalcaemia and
endanger renal function.
Features suggesting a less favourable outlook include:
1. Age> 40 years,
2. Afro-Caribbean ethnicity,
3. Persistent symptoms for more than 6 months,
4. the involvement of more than three organs,
5. lupus pernio
6. And a stage III/IV chest X-ray.
•
In patients with severe disease methotrexate (10-20 mg/week), azathioprine
(50-150 mg/day) and specific TNF-α inhibitors have been effective.
•
Chloroquine, hydroxychloroquine and low-dose thalidomide may be useful in
cutaneous sarcoid with limited pulmonary involvement.
•
Selected patients may be referred for consideration of single lung
transplantation.
•
The overall mortality is low (1-5%) and usually reflects cardiac involvement or
pulmonary fibrosis.