1
Fifth stage
Pediatric
Lec-8
.د
أوس
22/2/2016
Autosomal recessive inheritance
Many hundred disorders resulting from this type of inheritance are known
An affected individual is homozygous for the abnormal gene, having inherited an abnormal
allele from each parent, both of whom are unaffected heterozygous carriers
For two carrier parents, the risk of each child, male or female, being affected is 1 in 4 (25%)
All offspring of affected individuals will be carriers
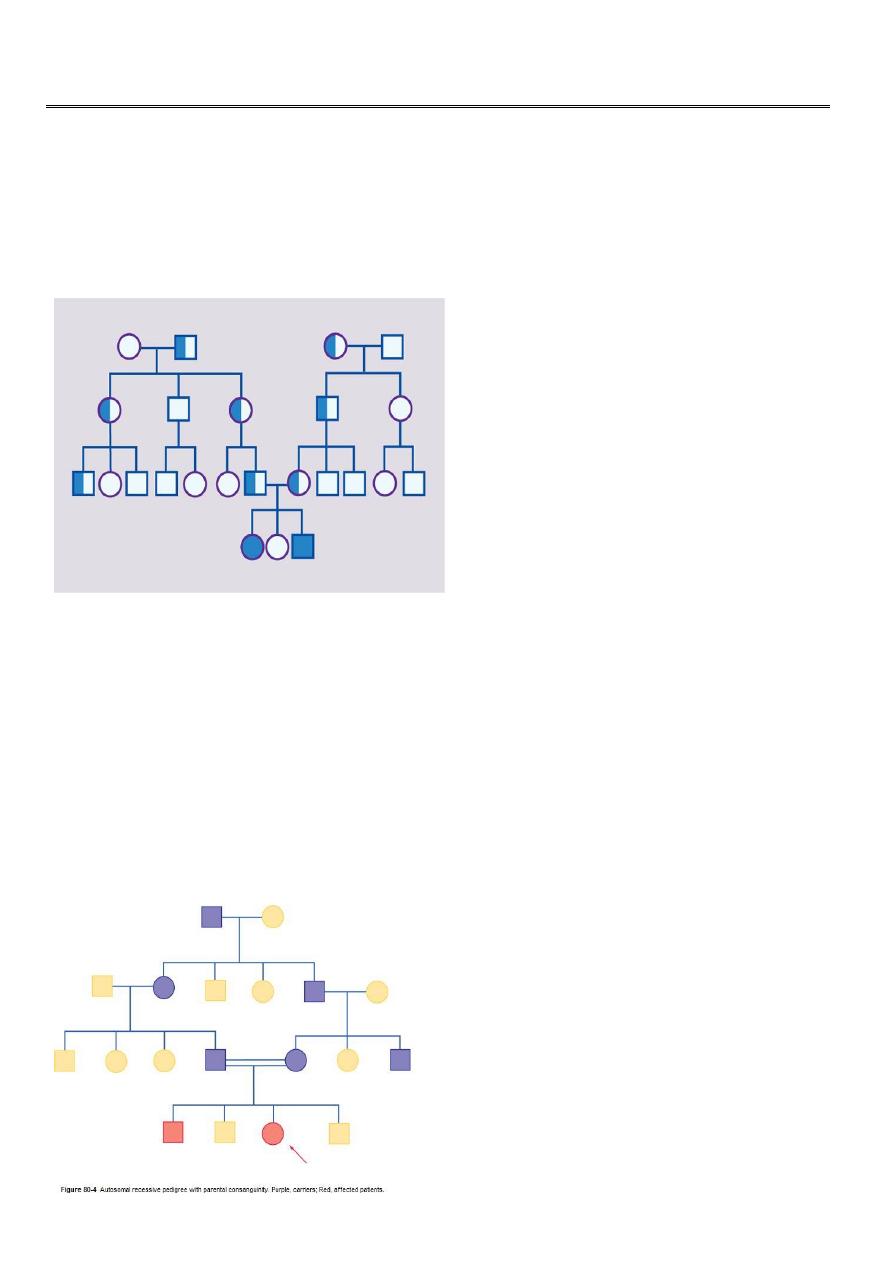
Consanguinity
It is thought that we all carry at least one abnormal recessive gene. Fortunately, our
partners usually carry a different one. Marrying a cousin or other relative increases the
chance of both partners carrying the same abnormal autosomal recessive gene, inherited
from a common ancestor. A couple who are cousins therefore have a small increase in the
risk of having a child with a recessive disorder

2
Racial factor:
Recessive gene frequencies may vary between racial groups
cystic fibrosis is common in north Europeans, sickle cell disease in black Africans and
Americans, thalassaemias in Mediterranean or Asian ethnicity and Tay-Sachs disease in
Ashkenazi Jews.
Inborn errors of metabolism:
Although individually rare, inborn errors of metabolism are an important cause of
paediatric morbidity and mortality. The specialised nature of the diagnostic tests and
subsequent management often means that these patients are managed in specialist
centres. However, as the prognosis for most patients depends upon the speed of diagnosis,
all doctors need to be familiar with their variable presentation and diagnosis.
Presentation:
An inborn error of metabolism may be suspected before birth from a positive family history
or previous unexplained deaths in the family
After birth, inborn errors of metabolism usually, but not invariably, present in one of five
ways

3
1. as a result of newborn screening, e.g. phenylketonuria (PKU), or family screening, e.g.
familial hypercholesterolaemia
2. after a short period of apparent normality, with a severe neonatal illness with poor
feeding, vomiting, encephalopathy, acidosis, coma and death, e.g. organic acid or urea cycle
disorders
3. as an infant or older child with an illness similar to that described above but with
hypoglycaemia as a prominent feature or as an ALTE (acute life-threatening episode) or
near-miss 'cot death', e.g. a fat oxidation defect such as medium-chain acyl-CoA
dehydrogenase deficiency (MCADD)
4. in a subacute way, after a period of normal development, with regression,
organomegaly and coarse facies, e.g. mucopolysaccharide disease or other lysosomal
storage disorder or with enlargement of the liver and/or spleen alone, with or without
accompanying biochemical upset such as hypoglycaemia, e.g. glycogen storage disease
5. as a dysmorphic syndrome.
Phenylketonuria:
It is either due to a deficiency of the enzyme phenylalanine hydroxylase (classical PKU) or in
the synthesis or recycling of the biopterin cofactor for this enzyme. Untreated, it usually
presents with developmental delay at 6-12 months of age. There may be a musty odour .
Many affected children are fair-haired and blue-eyed and some develop eczema and
seizures. Fortunately, most affected children are detected through the national biochemical
screening programme (Guthrie test).
Treatment of classical PKU is with restriction of dietary phenylalanine, whilst ensuring there
is sufficient for optimal physical and neurological growth. The blood plasma phenylalanine
is monitored regularly. The current recommendation is to maintain the diet throughout life.
This is particularly important during pregnancy, when high maternal phenylalanine levels
may damage the fetus
Galactosaemia:
This rare, recessively inherited disorder results from deficiency of the enzyme galactose-1-
phosphate uridyl transferase, which is essential for galactose metabolism
Presentation:
When lactose-containing milk feeds such as breast or infant formula are introduced,
affected infants feed poorly, vomit and develop jaundice and hepatomegaly and
hepatic failure

4
Chronic liver disease, cataracts and developmental delay are inevitable if the condition
is untreated.
Treatment:
Management is with a lactose- and galactose-free diet for life. Even if treated early, there
are usually moderate learning difficulties (adult IQ 60-80).
Glycogen storage disorders:
These mostly recessively inherited disorders have specific enzyme defects which prevent
mobilisation of glucose from glycogen, resulting in an abnormal storage of glycogen in liver
and/or muscle. There are nine main enzyme defects
The disorder may predominantly affect muscle (e.g. types II, V), leading to skeletal muscle
weakness. In type II (Pompe's disease) The heart is severely affected, leading to death from
cardiomyopathy. In other types (e.g. I, III) the liver is the main organ of storage, and
hepatomegaly and hypoglycaemia are prominent
Management is to maintain blood glucose by frequent feeds or by carbohydrate infusion
via a gastrostomy or nasogastric tube in infancy. In older children, glucose levels can be
maintained using slow-release oligosaccharides (corn starch